
Table of Contents
One goal of the U.S. Bureau of Mines metallurgy research program is to reduce the objectionable impact of mineral-processing operations upon the environment to the lowest possible level consistent with a viable minerals economy. The citrate process for flue gas desulfurization of (FGD) is a product of continuing Bureau research in pursuit of this goal, and the analytical work described in this paper was developed to support citrate process research.
The citrate process for flue gas desulfurization (FGD) is a product of continuing research by the U.S. Bureau of Mines to meet the goal of minimizing the objectionable effects of minerals industry operations upon the environment. The reduction of SO2 in solution by H2S to produce elemental sulfur by the citrate process is extremely complex and results in solutions that contain at least nine different sulfur species. Process solution analysis is essential to a clear understanding of process chemistry and its safe, efficient operation. The various chemical species , the approximate ranges of their concentrations in citrate process solutions, and the analytical methods evolved to determine them are hydrogen sulfide (-0M to 0.06M) by specific ion electrode, polysulfides (unknown) by ultraviolet (UV) spectrophotometry, elemental sulfur (-0M to -0.001M dissolved, -0M to -0.1M suspended) by UV spectrophotometry, thiosulfate (-0M to -0.25M) by iodometry or high performance liquid chromatography (HPLC), polythionates (-0M to -0.01M) by thin layer chromatogrphy (TLC), dithionite (searched for but not detected in process solutions) by polarography or TLC, bisulfite (-0M to 0.2M) by iodometry, sulfate (-0M to LM) by a Bureau-developed gravimetric procedure, citric acid (-0M to 0.5M) by titration or visible colorimetry, glycolic acid (-0M to 1M) by HPLC, sodium (-1 .5M) by flame photometry, and chloride by argentometric titration.
The chemical reactions occurring in citrate process operation will be described in a subsequent paper. Briefly, the citrate process comprises the following four elements:
1. SO2 absorption.-Sulfur dioxide is absorbed from cooled and cleaned flue gas by contacting the gas with a solution of citric acid buffered at about pH 4.5. In this step, SO2 absorption lowers the solution pH to about 4.
2. Absorbent regeneration.-High-purity elemental sulfur is precipitated by treating the SO2-rich absorbent solution with gaseous H2S. This restores the solution pH to 4.5, and permits the solution to be recycled for further absorption.
3. Sulfur separation.—Elemental sulfur precipitated during the regeneration step is separated from the solution by flotation. It is freed from occluded or entrained absorbent solution by autoclave melting at about 135° C. This permits withdrawal of pure molten sulfur from the lower phase in the autoclave. Supernatant absorbent from the upper phase, together with clarified absorbent from the flotation step, is then recycled for further SO2 absorption.
4. H2S generation.-Approximately two-thirds of product sulfur is used for H2S generation by reacting it catalytically at about 650° C with steam and a reducing agent such as natural gas, carbon monoxide, or methanol. The balance is cast into a form suitable for sale, storage, or shipment.
Elemental sulfur formation from H2S and SO2 in aqueous solution (“Wackenroder’s solution,” after the German apothecary who first reported it) is a phenomenon known since 1845; however, the chemistry of the process is extremely complicated and poorly understood. At least nine sulfur species are known to exist in solution during this reaction, and others have been postulated. Thus, citrate process research required the capability to analyze known sulfur compounds and to detect unknown compounds possibly present in process solutions. Methods for analyzing buffer species used in the process were also required, and are described herein.
The complexity of sulfur chemistry encountered in this research was compounded by the inconsistent nomenclature used by different authors. For example, an aqueous solution of sulfur dioxide has been called variously sulfurous acid, sulfite, bisulfite, or simply SO2. Similarly, the sulfur acid anions having the structure O3 S-So-SO3 2- have been called polythionic acids or polythionates most commonly; however, a prominent sulfur chemist recently advocated the name “polysulfanedisulfonate”. By this convention, the compound O3S-SSS-SO3 2-, historically called pentathionate, would be called tri- sulfanedisulfonate. In the present paper each sulfur compound is identified by the name that most accurately describes its chemical form as it is encountered in the citrate process. Thus SO2 dissolved in a pH 4 to 4.5 buffered absorbent is called bisulfite (HSO3-) rather than sulfite (SO3 2-), which is essentially absent at this pH, or sulfurous acid, which is nonexistent. The traditional polythionate nomenclature is used rather than “polysulfanedisulfonate.”
While the latter name may be technically more descriptive, it is unwieldy and is generally unfamiliar.
For easy reference, the paper is generally organized by compound. Sulfur compounds are discussed in order of increasing formal oxidation state. Fore the sake of completeness, brief descriptions of published procedures used are included together with those developed by the Bureau during the research.
Description of Samples
Citrate process absorbent is typically 0.5M (molar) in citric acid. In addition, because thiosulfate rapidly accumulates in the absorbent during process operation, fresh absorbent is made 0.25M in sodium thiosulfate. Thus, other sample constituents must be determined against this high background concentration of citric acid and thiosulfate. Recirculated absorbent is pale yellow and has a specific gravity of 1.1. Samples frequently contain elemental sulfur as a second phase, either as large flocs or a finely divided suspension. These samples can usually be clarified by filtration, although it is often necessary to use membrane filters with nominal pore size less than 1 micrometer to accomplish this.
Finally, samples typifying those from citrate process streams were known to contain at least the following solutes, at the concentrations shown where known: Sodium (1.5M), citric acid (0.5M), thiosulfate (0.25M), polythionates (So O6 2-, with n = 3 to at least 6), sulfate (0M to -1M), hydrogen sulfide (0M to 0.06M), bisulfite (0M to 0.2M), and elemental sulfur (-0M to -0.1M).
Analytical Methods
Sulfur Species
Hydrogen Sulfide
Hydrogen sulfide was analyzed with an Orion model 94-16 sulfide specific ion electrode and model 407 specific ion meter according to the manufacturer’s instructions. Only mercury and silver are reported to interfere with this measurement, and no interference was observed from sample components during this research. Because samples often contain elemental sulfur, they were filtered through a glass fiber filter disk (2 micrometers nominal pore size) prior to analysis. This precaution was taken to avoid accumulating sulfur on the electrode and not because the sulfur interfered with the analysis. The electrode is claimed by the maker to be sensitive to 10-~M sulfide. It was used routinely for analysis of H2S from 10 parts per million to 2,000 parts per million in process solutions.
Polysulfides
It is not believed possible to analyze individual polysulfide ions (HSn- or Sn 2-) in citrate process solutions because of the complex mixture of other labile sulfur ions present. Several titrimetric methods have been published that use iodine or cyanide to quantify polysulfides, but these could not be expected to succeed because of the reactivity of thiosulfate , polythionates , and elemental sulfur toward these same reagents. To avoid the foregoing difficulty, spectrophotometry was used for sulfide analysis.
Tetrasulfide and pentasulfide absorb light near 400 nanometers, with molar absorptivities , ε, of 1,140 and 2,000, respectively. The yellow color of recycled absorbent suggested that such ions might be present, but spectrophotometric scans of process solutions failed to reveal reproducible evidence for polysulfides. The method used would have detected tetrasulfide or pentasulfide at about 10- M.
Equipment and Material
A Cary model 15 UV-visible recording spectrophotometer was used to scan test solutions in fused quartz cells having either 1- or 10-millimeter path lengths.
Procedure
Samples were filtered to remove elemental sulfur and scanned from about 600 nanometers to about 200 nanometers. In some samples, but not all, the spectrum showed an absorbance maximum at 388 nanometers, suggesting polysulfide absorbance. Paradoxically, although all recycled citrate absorbent acquires a yellow color, only a few isolated test samples exhibited the absorbance at 388 nanometers. Most samples showed no absorbance maximum in this region. The yellow color of these samples was due to a very weak (0.01 to 0.05 absorbance units for a 10-millimeter path cell) general absorbance of unknown origin extending into the visible region above 400 nanometers.
Elemental Sulfur
Moderate concentrations of solid elemental sulfur (greater than about 1 to 5 grams per liter) were filtered and weighed; however, elemental sulfur is also present either dissolved or dispersed in unfilterable form. To analyze sulfur in these forms, a spectrophotometric method devised at the research center was used. This method uses as the basis for the determination the ultraviolet absorbance maximum exhibited at 268 nanometers by a trichloroethylene solution of sulfur.
Equipment and Material
Absorbance measurements were made on the Cary model 15 spectrophotometer. For the standard curve, 1-millimeter quartz cells were used. All other measurements were in 10-millimeter cells. Samples were shaken for extraction on a Burrell wrist-action shaker. Elemental sulfur (99.9 percent) was obtained from Montana Sulfur Co. Reagent-grade trichloroethylene (TCE) was obtained from Mallinckrodt. All other chemicals were the highest purity available commercially.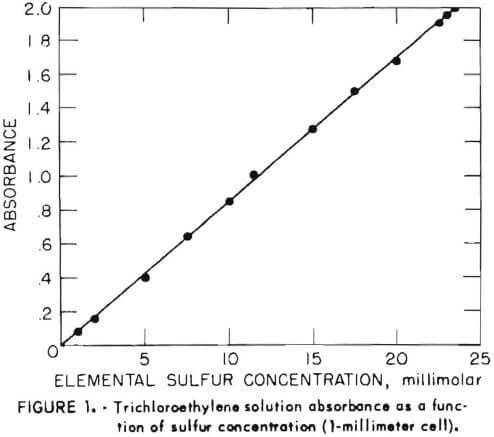
Procedure
The aqueous sample solution (10.0 milliliters) was contacted in a 60-milliliter separatory funnel with 10.0 milliliters of TCE by shaking for 20 minutes on the wrist-action shaker. After phase disengagement, the optical absorbance of the TCE solution was measured at 268 nanometers. Figure 1 shows the absorbance of solutions containing varying amounts of pure elemental sulfur. The molar absorptivity of sulfur under these conditions is 850.
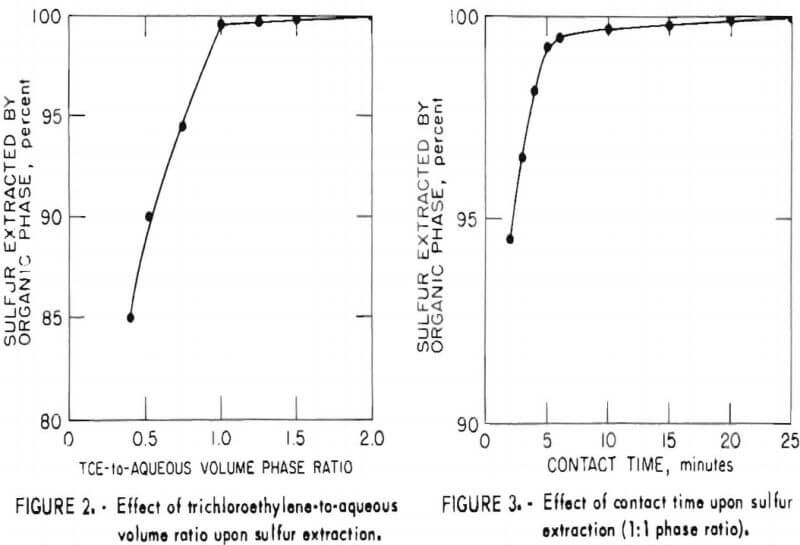
In order to establish optimum conditions for the analysis, sulfur extraction was determined as a function of phase ratio and contact time as shown in figures 2 and 3, respectively. As may be seen from these figures, the small incremental improvement in sulfur extraction obtainable by increasing the TCE-to-aqueous ratio above 1 would be offset by an attendant decrease in sensitivity for the more dilute TCE solution. Similarly, no significant benefit would be gained by a contact time greater than 20 minutes.
Tests were run to determine whether other ions present in typical citrate process samples would interfere with this analysis. No interference was observed for solutions 0.5M in citric acid and also containing 0.2M sulfide , thiosulfate, bisulfite, trithlonate, tetrathionate, or dithionite.
The sulfur concentration in the aqueous is calculated from the equation,
C = A/850 b,
where C = sulfur concentration in moles per liter,
A = solution absorbance in absorbance units,
and b = cell path length in centimeters.
Samples for which the absorbance exceeds the instrument scale may be diluted with TCE without affecting the determination, in which case a correction for dilution must be applied to the relationship above.
Thiosulfate
Two methods were used for thiosulfate determination. In most cases standard iodometric titration was suitable. (See also “Bisulfite” section of the present report). Interference by bisulfite was prevented by masking with formaldehyde; a correction to the Iodine titer must be applied for hydrogen sulfide present in the sample.
During investigations of citrate process chemistry and reactions, a thiosulfate analysis was required that was not susceptible to error from unknown components of sample solutions that might consume iodine, thereby introducing an indeterminate error in the iodine titer. A high-performance liquid chromatographic (HPLC) procedure was developed to supplement the iodometric method.
Equipment and Material
A Perkin-Elmer model 1220 liquid chromatograph and model LC55 variable wavelength UV detector coupled to a Beckman model 1005 strip chart recorder were used for the analysis. The sample injection valve was fitted with a 30- microliter sample loop. The analytical column was 2.6-millimeter-inside-diameter by 0.5-meter-long stainless steel packed with Zipax strong anion exchange pellicular resin (DuPont Instrument Co.). Peak areas were integrated electronically by a Spectra-Physics Autolab System IV data analyzer. Reagent-grade cadmium sulfate in degassed distilled water was used as the eluent (mobile phase).
Procedure
Prior to injection onto the column, samples were diluted fiftyfold with distilled water to prevent overloading the low-capacity resin. Samples were eluted with a solution of 0.38 gram cadmium sulfate per liter of degassed distilled water at a flow rate of 1.5 milliliters per minute (1,000 pounds per square inch pressure). Figure 4 shows the analysis of regenerated absorbent solution containing 0.25M thiosulfate in 0.5M citric acid buffer. The significant feature of this analysis is that thiosulfate, which elutes first among the components of typical process solutions, is separated from citric acid. By all other HPLC procedures screened during development of this procedure, citric acid and thiosulfate eluted together, making thiosulfate determination impossible. The thiosulfate retention time of 7.7 minutes is reproducible to within 0.3 minute over the range from 0.1M to 0.25M.
Polythionates
Polythionates , SnO6 2- with n≥3, are always present in citrate process solutions. Dithionate, S2O5 2-, which is structurally similar, is chemically distinct from higher members of the series and was not detected in process solutions. The detection limit for dithionate by methods described herein is estimated 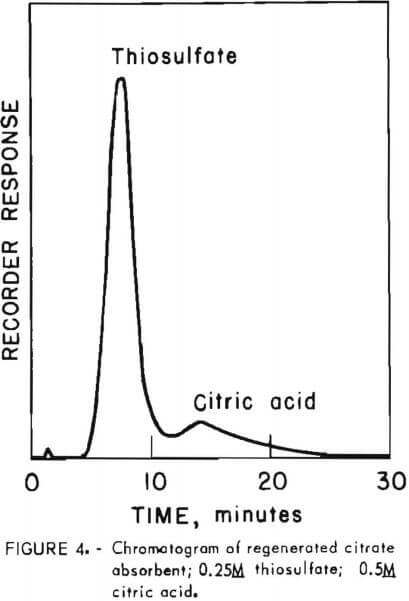 to be 50 parts per million. Although several methods have been reported for analyzing individual polythionates, none is satisfactory for large numbers of samples. Kurtenacker and Goldbach determined polythionates through S5O6 2- in mixtures by titrating five aliquots of each sample. Jay published a procedure for determining total but not individual (polythionates in a sample; however, this method was not reliable in the present study, possibly because of interference from the much higher concentrations of other solutes. Chapman and Beard reported a HPLC method for individual polythionates, but this method requires high sample dilution, and polythionates are not detected below about 1 to 2 grams per liter in the original sample. Polythionate analysis by thin-layer chromatography (TLC) was found to be a rapid and convenient procedure compatible with citrate process research needs.
to be 50 parts per million. Although several methods have been reported for analyzing individual polythionates, none is satisfactory for large numbers of samples. Kurtenacker and Goldbach determined polythionates through S5O6 2- in mixtures by titrating five aliquots of each sample. Jay published a procedure for determining total but not individual (polythionates in a sample; however, this method was not reliable in the present study, possibly because of interference from the much higher concentrations of other solutes. Chapman and Beard reported a HPLC method for individual polythionates, but this method requires high sample dilution, and polythionates are not detected below about 1 to 2 grams per liter in the original sample. Polythionate analysis by thin-layer chromatography (TLC) was found to be a rapid and convenient procedure compatible with citrate process research needs.
Equipment and Material
High-surface-area silica gel plates (0.2 millimeter thick) equivalent to Merck 5775 are most satisfactory. A Kotes System I densitometer with baseline corrector was used for quantifying developed plates. A Soltec model 281 strip-chart recorder was used to convert densitometer output to optical density spectra. Peaks were integrated with the Spectra-Physics Systems IV data analyzer. Reagent-grade chemicals were used in distilled, deionized water to prepare eluent solutions. Potassium trithionate and pentathionate were synthesized by the method of Goehring, Stamm, and Feldman. Sodium tetrathionate was purchased from ICN Pharmaceuticals, Inc.
Procedure
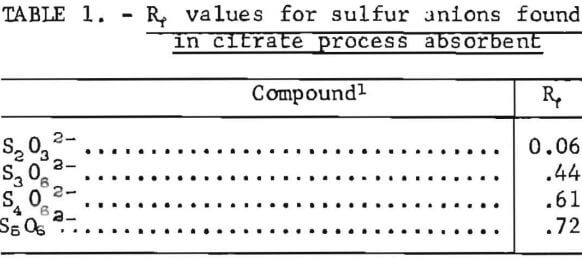
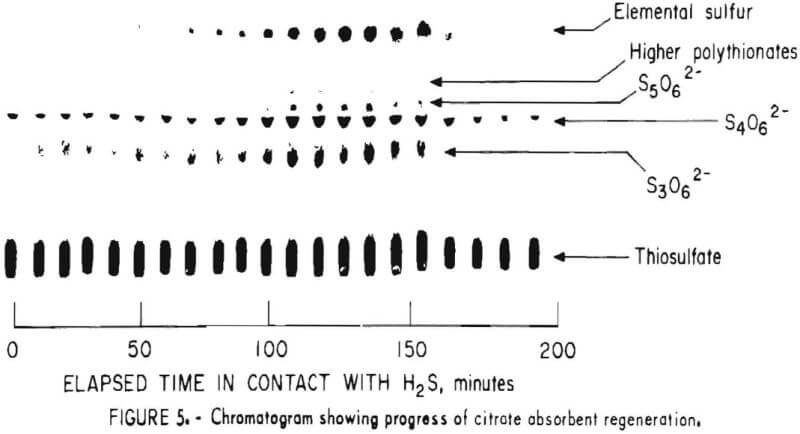
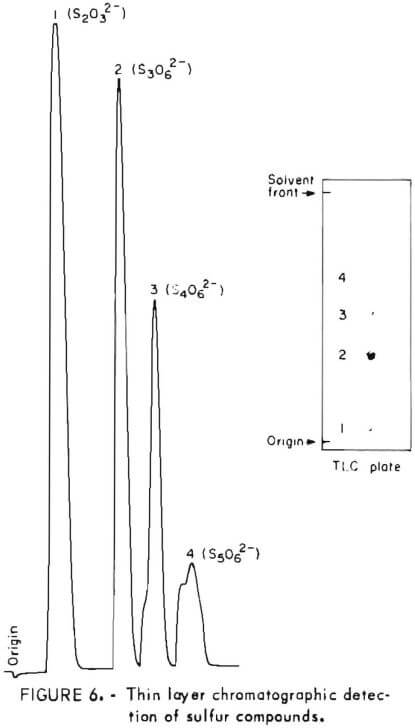
Samples were diluted 1:50 with distilled water prior to chromatography. Those samples containing solid sulfur were filtered prior to analysis to avoid plugging the spotting syringe. Chromatograms were developed by ascending solvent consisting of ethanol, p-dioxane, concentrated ammonia, water (12:4:2:1). Developed chromatograms were visualized by spraying with either ethanolic silver nitrate to aqueous palladium chloride acidified with a few drops of HCl in 50 milliliters of solution. Silver nitrate is the more sensitive reagent; however, plates sprayed with silver nitrate darken after 1 or 2 hours and are unsuitable for densitometry. Plates visualized with palladium chloride remain essentially unchanged and may be quantified at any time after spraying. Figure 5 illustrates the TLC analysis of SO2-loaded absorbent during regeneration with H2S. This plate was visualized with silver nitrate. Figure 6 shows the separation and corresponding densitometer scan of 3 microliters of a synthetic mixture containing thiosulfate (spot 1, 7.99 grams per liter), trithionate (spot 2, 3.58 grams per liter), tetrathionate (spot 3, 1.26 grams per liter), and pentathionate (spot 4, 0.77 gram per liter). The silica gel plate was a commercial product on a transparent plastic backing that permitted operation of the densitometer in the visible transmission mode. Spot 4 corresponding to pentathionate was too faint to photograph satisfactorily; nevertheless, quantification was possible. By this method, polythionate concentrations at least as low as 100 parts per million may be analyzed. Table 1 lists the R, values for thiosulfate and the polythionates analyzed in process solution by this method.
Dithionite
 Dithionite (S.O. 2-), a compound of S, was regarded as a possible intermediate in the reduction of SO2 in the citrate process. It is not separated from the known components of process solutions by the TLC solvent system above; however, a solvent consisting of methanol, n-butanol, water (3:1:1) separates dithionite and thiosulfate with Rf value; of 0.80 and 0.64, respectively. Dithionite may also be visualized by spraying with either ethanolic silver nitrate or aqueous palladium chloride. Extensive examination of process solutions failed to find dithionite under conditions that should have detected about 50 parts per million or less.
Dithionite (S.O. 2-), a compound of S, was regarded as a possible intermediate in the reduction of SO2 in the citrate process. It is not separated from the known components of process solutions by the TLC solvent system above; however, a solvent consisting of methanol, n-butanol, water (3:1:1) separates dithionite and thiosulfate with Rf value; of 0.80 and 0.64, respectively. Dithionite may also be visualized by spraying with either ethanolic silver nitrate or aqueous palladium chloride. Extensive examination of process solutions failed to find dithionite under conditions that should have detected about 50 parts per million or less.
Bisulfite
Bisulfite (HSO3-) was determined by a standard iodometric method. Because thiosulfate interferes with this titration, it is necessary to titrate two aliquots of sample, one of which is pretreated with formaldehyde to sequester bisulfite as its addition compound with formaldehyde.
A suitable aliquot of sample (2 to 10 milliliters) is added to about 50 milliliters of water. Formaldehyde (5 milliliters of 37-percent-HCHO solution) is added, followed by sufficient NaOH (-1M) to make the solution aklaline to phenolphthalein. The solution is allowed to stand for 10 minutes to allow complete reaction of dissolved SO2 with the formaldehyde.
A second aliquot of sample (2 to 10 milliliters) is added in one portion to an excess of standard iodine solution (-0.1N) contained in about 50 milliliters of water. The volume of iodine solution is recorded as “A.” The sample is acidified with 2 milliliters of glacial acetic acid and titrated with standard thiosulfate solution (-0.1N) to a faint yellow color. Starch indicator is then added, and titration with the standard thiosulfate solution is continued to a colorless end point. The total volume of thiosulfate is recorded as “B.”
Titration of the first sample aliquot is now completed by adding 2 milliliters of glacial acetic acid to the solution, followed by starch indicator and then titrating with standard iodine solution (-0.1N) to the blue end point. The volume of standard iodine solution is recorded as “C.”
Analyte concentrations, in grains per liter, are calculated as follows:
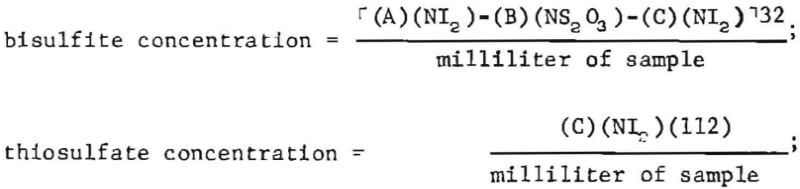
where NI2 = normality of standard iodine solution,
and NS2O3 = normality of standard thiosulfate solution.
Iodine solution is standardized against arsenious oxide (As2O3) as either the dry reagent or standard solutions available from chemical suppliers. The thiosulfate solution is then standardized against the standard iodine.
Sulfate
A gravimetric method developed by the Kraft paper industry was adapted by the Bureau for use with citrate process samples. Although simplified, it does not sacrifice accuracy for samples containing from 0.05 to 50 grams per liter of sulfate, which represents the range of concentrations encountered.
Equipment and Material
An IEC model K centrifuge was used for separating the first BaSO4 precipitate. Carbon dioxide used for deoxygenating solutions was obtained in cylinders from commercial vendors. All chemicals were reagent grade. Iodine reagent (about 0.3N) for selectively oxidizing thiosulfate to tetrathionate was prepared by dissolving 38.1 grams iodine and 120 grams potassium iodide in about 50 milliliters water and diluting to 1 liter after complete dissolution. Distilled water (33 to 500 milliliters) was deoxygenated by bubbling CO2 through it for 5 minutes and was then stored in a tightly stoppered bottle. Two-percent solutions of HCl and NaOH were deoxygenated in the same way. (Some carbonate forms in the NaOH solution; however, this does not interfere with the analysis.)
Procedure
The analysis is based on the classical precipitation of sulfate as the barium salt; however, because of the high concentrations of solutes other than sulfate in process samples and the low solubility of other barium salts such as citrate, samples were analyzed by the following four-step method:
1. Choose an aliquot of sample to contain 1 to 125 milligrams of sulfate. Pipet the aliquot into a 150-milliliter beaker and add 5.0 milliliters of 37- percent-formaldehyde solution and 5.0 milliliters of isopropyl alcohol. Dilute to 40 milliliters with deoxygenated water and adjust the pH to 4.0 with deoxygenated HCl or NaOH as required. Allow the sample to stand for 5 minutes to insure complete reaction of bisulfite and formaldehyde. (This will prevent oxidation of bisulfite to sulfate during addition of iodine in the next step.) During this interval, purge oxygen from the 0.3N iodine solution by bubbling CO2 through it. Add iodine reagent to the sample to a blue starch indicator end point, then add 1 milliliter excess. This converts thiosulfate quantitatively to tetrathionate.
2. Add about 5 grams of lump-free ammonium chloride to a 250-milliliter centrifuge bottle, displace air in the bottle with CO2, and add the sample from step 1, washing the sample from the beaker with deoxygenated water. Adjust sample volume to about 175 milliliters. Swirl the stoppered centrifuge bottle until the ammonium chloride dissolves, then add 1.25 to 1.5 grams solid BaCl2·2H2O and swirl thoroughly. Centrifuge at 2,000 revolutions per minute for 30 minutes. Repeat if necessary to clarify the supernatant. When a clear supernatant is obtained, decant and discard it.
3. Transfer the precipitate from step 2 quantitatively into a 400-milliliter beaker. Dilute with water to a volume of about 400 milliliters, and add 2 milliliters of 2-percent HCl. Heat on a hotplate at medium heat for 30 minutes, then add 10 milliliters concentrated HCl, cover with a watch glass and boil for 2 minutes. Remove the beaker from the heat, add methyl orange indicator, neutralize with concentrated aqueous ammonia, reacidify with 10 drops of excess concentrated HCl, and boil for 5 minutes. Remove from heat and cool overnight. Filter the supernatant solution through filter paper equivalent to S. and S. No. 589 containing 5 milliliters pulp. (Do NOT transfer the bulk precipitate to the filter paper.) Wash down the sides of the beaker containing the BaSO4 precipitate with 20 milliliters concentrated nitric acid and 20 milliliters concentrated perchloric acid. Place the filter paper and pulp into this beaker and digest to heavy fumes over medium heat.
If the residue is colored, add concentrated nitric acid and continue fuming until colorless. Finally, fume down to a 2-milliliter volume, cool, and add 250 milliliters distilled water, methyl orange indicator, neutralize with aqueous ammonia, and reacidify with 6 drops excess concentrated HCl.
4. Heat the sample, add 5 milliliters of a solution of 225 grams BaCl2·2H2O in 2 liters of water. Cover the sample and boil for 5 minutes , then allow to simmer for 30 minutes. Remove from heat and cool overnight. The precipitate can now be recovered by filtration, after which BaSO4 is determined by heating the filter paper plus precipitate at 900° C for 30 minutes in an annealing cup and weighing the BaSO4.
Buffer Species
Citric Acid
For process control and material balance calculations, it is essential to be able to quantify the buffer component of process solutions as well as the reactants. The following procedures were used.
Equipment and Material
Automatic titrations were performed on a Mettler DK10-DK11 titrimeter with model GA10 recorder. Visible absorbance was measured on the Cary model 15 spectrophotometer. All chemicals were reagent grade.
Procedure
A Bureau-developed titrimetric method was used for most laboratory experiments and for pilot plant testing under this program. The basis for the method is the release by ordinarily tribasic citric acid of a fourth acidic hydrogen upon coordination with cupric ion. A 1.0-milliliter aliquot of sample is diluted to about 50 milliliters with water. Hydrogen peroxide (5 milliliters of 30-percent solution) is added, and the solution is heated to 90° C for 5 minutes to destroy bisulfite, which interferes with the titration. After cooling, the sample is titrated to just past the end point with standarized NaOH solution, about 0.1N. (The titrimeter should be operated in the differential mode to provide sharp definition of the end points.) Copper sulfate penta-hydrate, about 0.6 gram, is then added, and the sample is titrated to a second end point. The volume of titrant added between the two endpoints is recorded. At the end of the titration the concentrations of citrate and cupric ion should be approximately 0.005M and 0.02M, respectively. The citric acid concentration (C), in grams per liter is calculated from
C = (N) (V) (192.1),
where N = normality of NaOH titrant,
and V = volume of titrant added between end points, in milliliters.
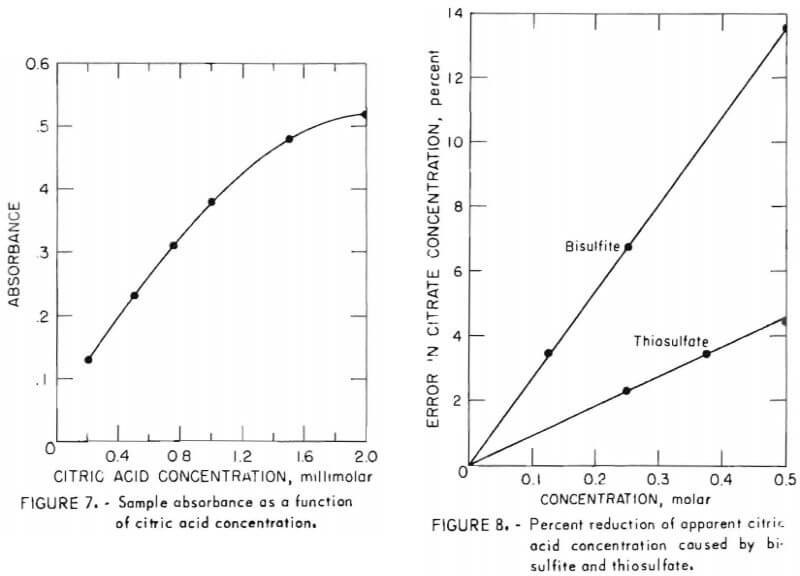
An alternate citric acid analysis used by the dairy industry has recently been found to be suitable for citrate process samples and to require less instrumentation and operator skill. In this procedure, 1.0 milliliter of sample containing not more than 1 micromole (192 micrograms) of citric acid is placed in a test tube, followed by 1.3 milliliters of pyridine. The solution is mixed by swirling. Acetic anhydride (5.7 milliliters) is added, after which the contents are swirled briefly, then incubated for 30 minutes in a constant temperature bath at 32° ±0.25° C. Citrate acid is then determined by measuring the absorbance of the resulting solution at 435 nanometers and comparing sample absorbance with n standard curve.
Because the author of this method stated that solution absorbance in this analysis does not obey Beer’s law exactly, absorbance was measured for a series of samples between 0.2 millimolar and 2 millimolar. Figure 7 shows absorbance as a function of citric acid concentration in this range. While the curve is not liniar, it is nearly so up to 1 millimolar, and satisfactory values are possible by comparing sample absorbance with such a standard curve even beyond 1 millimolar.
To determine whether other potential components of sample solutions would interfere with this analysis, it was repeated for solutions containing 0.02M Fe3+, Cu2+, Ni2+, Cr3+, Sn2+, Ca2+, Mg2+, Al3+, Zn2+, Na+, Cl-, or SO4 2- in 0.1M
citric acid. No interference was seen; however, the following precautions must be observed when using this analysis:
- Both bisulfite and thiosulfate depress sample absorbance. Figure 8 shows the percent by which the apparent concentration of known 0.5M citric acid is reduced by varying amounts of these solutes. Because this effect is linear as shown, a correction may be applied to the apparent citric acid analysis when these components are present in the sample. A correction curve such as figure 8 should be prepared for each system under study; nevertheless, error due to bisulfite or thiosulfate is not believed to be a serious impediment to use of this procedure. At the greatest concentrations of these solutes usually encountered, 0.15M and 0.25M for bisulfite and thiosulfate, respectively, the corresponding errors are 4 percent and 2.3 percent. Furthermore, only absorbent freshly contacted with SO2 contains such a high concentration of bisulfite. At all other points in the system the bisulfite concentration is below about 2 grams per liter, at which level the corresponding error is below 1 percent.
- Samples must be diluted carefully in volumetric glassware to reduce the citric acid concentration to about 0.001M prior to adding pyridine.
- Accurate citric acid results are obtained for samples with pH below 4.0; however, above pH 4.0, the apparent concentration is low as shown in figure 9. This effect is easily prevented by adjusting sample pH to 4 or lower prior to diluting and adding pyridine.
- Both pyridine and acetic anhydride are degraded by light and/or moisture. Fresh reagent should be used daily, and stock reagent should be stored dry and in low actinic glass containers.
Glycolic Acid
Glycolic acid (hydroxyacetic acid) was successfully tested as a possible lower cost alternative to citric acid buffer in the citrate process. It may be analyzed as the methyl ester by gas chromatography; however, this is a relatively time-consuming procedure. A simple HPLC method has been found to be suitable. Although no exhaustive work has been done on this analysis, the preliminary results reported here show that HPLC can be used to separate and quantify glycolic acid in citrate process absorbent.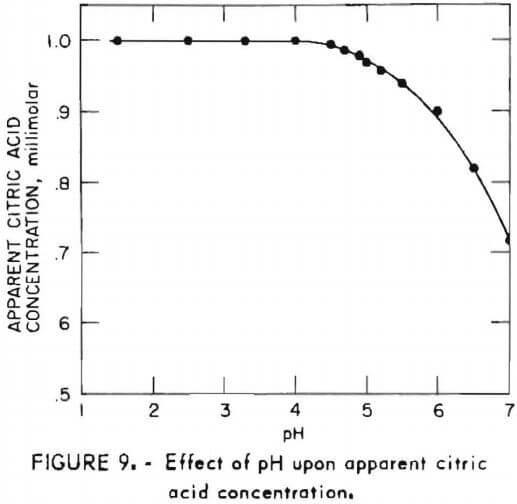
Equipment and Material
The liquid chromatograph and associated equipment were as described for thiosulfate analysis. The analytical column was 2.6-millimeter-inside-diameter by 0.5-meter-long stainless steel cleaned and packed as described by Kirkland with Permaphase AAX weak anion exchange resin (DuPont).
Procedure
A 30-microliter sample loop was used to apply samples to the column. Elution was by reagent-grade sodium sulfate (7 x 10 -3 M) in degassed distilled water at a flow rate of 1 milliliter per minute (1,000 pounds per square inch). Sample components were detected by UV absorbance at 205 nanometers.
Figure 10 shows the analysis obtained in a 1.0M solution of glycolic acid containing 0.6M sodium thiosulfate. As with citric acid, many eluents failed to separate the glycolic acid buffer from thiosulfate, which is always found in process solutions. Sodium sulfate eluent provided good resolution of the glycolic acid and thiosulfate peaks. The method is limited by the low UV absorptivity of glycolic acid; however, it has been used for solutions as low as 0.05M. To avoid overloading the low-capicity resin, samples were diluted with water prior to injection. For the analysis of figure 10, a 500-fold dilution was used. Less dilution may be used to improve the sensitivity for glycolic acid if the concentrations of other solutes are found to be lower than the 0.6M thiosulfate in the analyte of figure 10.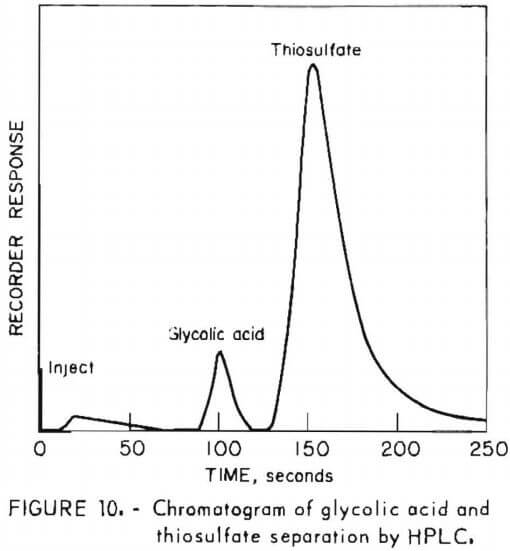
Miscellaneous Species
Sodium
Sodium ion was analyzed by flame photometry. An Instrument Laboratory
Inc. model 343 photometer was used with continuous Internal lithium standard. The limit of detection by this method is 1 part per million.
Undiluted citrate process solutions are sufficiently viscous that precautions must be taken to insure that the sample aspiration rate is comparable to that of the sodium standard solutions. This may be accomplished by either (1) diluting the sample prior to analysis , if the sodium concentration after dilution remains above the detection limit (process samples were routinely diluted 100:1 with water) or (2) adding citrate to standard solutions at a concentration equal to that of the sample.
Chloride
Process solutions were analyzed for chloride ion by an adaptation of the Volhard method. Because sulfide and sulfite interfere with the analysis, a preliminary treatment with is necessary to oxidize sulfite to sulfate followed by boiling to decompose excess H2O2, and to expel residual sulfide.
In a typical analysis, an aliquot of sample containing about 5 to 100 milligrams of chloride is placed in a Erlenmeyer flask and diluted with water to about 100 milliliters. About 1 milliliter of 30-percent-H2O2 solution is added, after which the flask is covered with a watch glass and boiled for about 5 minutes. After cooling, an excess of standard AgNO3 (about 0.1N) is added; then, the sample is acidified with 2 milliliters of concentrated nitric acid. Iron(III) indicator (2 milliliters of saturated FeNH4(SO4)2 solution) is added followed by 5 milliliters of chloride-free nitrobenzene. After the flask is vigoriously shaken, the excess AgNO3 is titrated with standard (0.1N) potassium thiocyanate solution to the first red FeSCN 2+ color that persists for 1 minute.
The chloride concentration in grains per liter, is then calculated from
Cl- = [ (A) (B) – (C) (D) ] (35.46)/E
where, A = milliliter of standard AgNO3 added,
B = normality of standard AgNO3,
C = milliliter of standard potassium thiocyanate used,
D = normality of standard potassium thiocyanate,
and E = milliliter of sample taken.
The foregoing procedure is satisfactory for solutions containing more than about 100 parts per million of chloride. In solutions containing less than 100 parts per million of chloride, a chloride-specific ion electrode is recommended.
General Methods
Direct-Current Polarography
One of the principal objectives of citrate process laboratory research was to identify the compounds of sulfur that exist in an operating system. In addition to TCL, direct-current (DC) polarography was used to screen process samples for previously unknown compounds.
Other investigators have used polarography to analyze thiosulfate, sulfide, trithionate, dithionite, and bisulfite, but the supporting electrolyte in these investigations was phosphate, acetate, nitrate, or chloride. Because samples generated in citrate process research are in relatively concentrated (0.5M) citric acid, this was used as supporting electrolyte for these tests, diluted to a concentration in the cell of 0.01M.
Equipment and Material
The polarographic cell consisted of a 1-liter, five-neck, round flask. The dropping mercury electrode was inserted in the central neck, nitrogen for deaerating the solution was admitted through the rear neck, the front neck accomodated the saturated calomel reference electrode, and the necks on either side of center were for two graphite auxiliary (‘’working”) electrodes. Potential to the cell was supplied by a Princeton Applied Research model 173 potentiostat and was varied by an Elscint model ABA-26 automatic baseline advance. Applied potential was displayed on a Hewlett-Packard model 3440A digital voltmeter. Current, in microamperes, was displayed on the potentiostat and recorded simultaneously on a Honeywell Electronic 19 recorder. The dropping mercury electrode was made from standard capillary bore glass tubing and was regulated to deliver 1 drop every 3 seconds.
Procedure
Ten milliliters of sample were diluted with distilled water to 1.0 liter in the cell, after which the solution was deaerated by bubbling nitrogen through it for 10 minutes. Because the half-wave potentials of dithionite and sulfide were indistinguishable below pH 7, the sample pH was adjusted to 7 prior to final dilution. At this pH, the known sulfur ions could be analyzed in mixtures such as process solutions. The solution was then analyzed by scanning between +350 millivolts and -1,000 millivolts at a rate of 200 milli-volts per minute. Table 2 lists the half-wave potentials and corresponding current measured for electroactive sulfur ions during these tests.
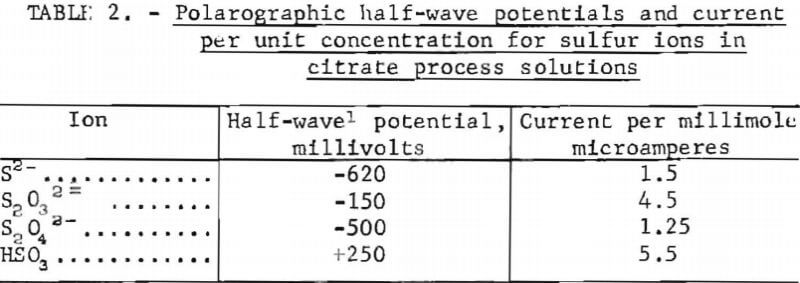
No polarographic wave was observed for sulfate or the polythionates. The main objective of the polarographic analyses was not to quantify the sulfur ions, which was done by other methods described, but to search for compounds not previously known; however, no reproducible evidence for the existence of previously undetected compounds was obtained.
Total Sulfur
The total sulfur content of test solutions was required both as a check of other analyses and for material balance calculations. It was determined by oxidizing all sulfur to sulfate, which was then determined gravimetrically as the barium salt.
Equipment and Material
Only standard laboratory glassware was required in addition to a magnetic stirrer-hotplate. For several simultaneous analyses a 230-volt hotplate having a 45- by 60-centimeter surface was used.
Procedure
Aliquots of sample (5.00 milliliters) in 250-milliliter beakers were diluted to about 50 milliliters with water. Bromine (about 2 milliliters) was added after which the sample was heated with magnetic stirring until no bromine droplets were visible. Concentrated nitric acid (5 milliliters) was added, and the solution was then boiled gently to a straw yellow color. Concentrated HCl (20 milliliters) was then added, and boiling was maintained to expel nitrates. Further 20-milliliter additions of HCl were made with continued boiling until the solution was colorless. The solution was then concentrated by boiling to a volume of about 25 to 50 milliliters, after which 15 milliliters of 0.25M BaCl solution was added. The solution was boiled gently for 10 minutes, allowed to cool, and filtered under gentle suction onto previously tared Whatman GF/C glass fiber disks. For routine purposes, predrying of the filter disks was not justified as they were found to contain less than 0.15 percent moisture by weight.
This relatively rapid and simple analysis produced replicate sulfur values typically within 2 percent or less.
Citrate process for flue gas desulfurization
The methods described herein comprise those analytical procedures that were evolved during the 7-year period in which the citrate process developed from preliminary laboratory experiments to a demonstration plant under construction to treat exit gas from a 60-mogawatt coal-buring powerplant. They should provide satisfactory results for other investigators studying sulfur chemistry as encountered in FGD systems, particularly of the regenerable type.
During preparation of this manuscript, an innovation Infrared spectrophotometric method for determining citric acid, thiosulfate, and sulfate simultaneously in scrubber solutions was published. This method was not studied during the present investigations, but it may be of interest to other researchers having access to a modern double-beam infrared spectrophotometer.