
Table of Contents
Zeolites are crystalline aluminosilicates of the alkaline and alkaline-earth metals. They possess many desirable ion- exchange, molecular sieving, and catalytic properties, which make them valuable mineral commodities. Synthetic zeolites have been used for over 25 yr in commercial processes, but only recently have natural zeolites been viewed as potentially valuable mineral commodities. Because of this interest, the Bureau of Mines undertook this report to discuss the occurrence, synthesis, mineralogy, economics, and uses of these versatile minerals.
Zeolites were first identified by Constedt in 1756. The name zeolite, from the Greek words meaning “boiling stone,” alludes to the frothing and bubbling observed by Cronstedt when he heated several crystals. In 1845, Way discovered that certain soils retained ammonium salts. Breck reported that hydrated silicates in the soil were found to be responsible and that these were probably the first ion-exchange experiments. Weigel and Steinhoff, in 1925, were the first to determine that chabazite selectively absorbed smaller organic molecules and rejected large molecules. This phenomenon was described by McBain in 1932 as “molecular sieving.” It was not until the 1940’s and 1950’s that research on the properties of zeolites increased dramatically, especially at the laboratories of Barrer at the Imperial College in London. Barrer’s work included the recognition of various molecular sieve types, quantitative studies of molecular sieving, and the use of ion exchange to modify the exchange properties of molecular sieves.
As knowledge of the properties of zeolites increased, it became apparent that they could be utilized for industrial processes. Zeolites, however, were considered to be mineralogical curiosities that filled vugs and fractures in igneous rocks. Large tonnages of natural zeolites were not discovered until the late 1950’s when Ames, Sands, and Goldich, Deffeyes, and Mumpton reported on vast sedimentary deposits in the Western United States. Prior to the 1950’s, the emphasis was on the synthesis of zeolites for commercial use.
The synthesis of zeolites was reported as early as 1862, although Breck noted that the early work was not substantiated by X-ray diffraction and some of it is not reproducible. He credited Barrer with the first synthesis of analcime-type zeolites substantiated by X-ray diffraction in 1951.
Initially, zeolites were synthesized under temperatures and pressures thought to be responsible for the crystallization of zeolites in basaltic rocks. Breck reports that in 1959, Milton and coworkers at Union Carbide Corp. developed a new technique that allowed low-temperature synthesis of zeolites. Their technique used extremely reactive components in a closed system and temperatures of crystallization more typical of those for organic compounds. This procedure was adaptable to large-scale production of synthetic zeolites.
By the 1980’s, 40 natural zeolites had been identified (table 1), over 100 zeolites had been synthesized, and natural zeolite resources within the United States were estimated to be 10 trillion st. Approximately 163,000 st of natural and synthetic zeolites were consumed in the United States in 1980. Of this, 5,000 st were natural zeolites. In 1985, approximately 13,000 st of natural zeolites were mined in the United States. While synthetic zeolites have been used extensively in commercial applications, demand for natural zeolites has been limited. Their primary use is in areas where the use of synthetic zeolites would be uneconomical. Several uses, both commercial and experimental, include ammonium-ion removal for aqua-culture and uranium mine waste water, odor control for chicken farming and cat litter, and removal of heavy metal ions from nuclear, mine, and industrial waste waters.
Definitions
Zeolites are crystalline-framework aluminosilicates based on a three-dimensional network of SiO4 tetrahedra, with all four oxygens shared by adjacent tetrahedra. Zeolites may be represented by the empirical formula M2/nO·Al2O3·xSiO2·yH2O. M is an alkali or alkaline earth cation of n valence, x is a number between 2 and 10, and y is a number between 2 and 7. The principle cations are sodium, potassium, magnesium, calcium, strontium, and barium. The cations are loosely bound in the structure and may be exchanged, to varying degrees, by each other. The framework contains channels and interconnected voids occupied by cations and water molecules. Most zeolites can be reversibly dehydrated.
Zeolites are characterized by the following properties:
- High degree of hydration.
- Low density and large void volume when dehydrated.
- Stability of the crystal structure of many zeolites when dehydrated.
- Cation exchange properties.
- Uniform molecular-sized channels in the dehydrated crystals.
- Various physical properties such as electrical conductivity.
- Adsorption of gases and vapors.
- Catalytic properties.
Molecular sieves are materials that, because of their internal structure, can selectively adsorb molecules according to their size and/or shape. All zeolites are molecular sieves, but not all molecular sieves are zeolites. Activated carbon, activated clays, alumina powder, and silica gels are examples of molecular sieves that are not zeolites.
A gel is a hydrous metal aluminosilicate that is prepared from either aqueous solutions, reactive solids, colloidal sols, or reactive aluminosilicates such as the residue structure of metakaolin (derived from kaolin clay by dehydroxylation) and glasses.
Structure and Classification
Zeolites are crystalline frameworks of oxygen, aluminum, and silicon extending in a three-dimensional framework. Figures 1A and 1B illustrate the basic structural building blocks. In both cases, either a silicon or aluminum atom is at the center of a tetrahedron formed by the oxygen atoms. If an aluminum atom is present rather than a silicon atom, a positive metal ion is required to maintain a charge balance (figs. 1B-1C).
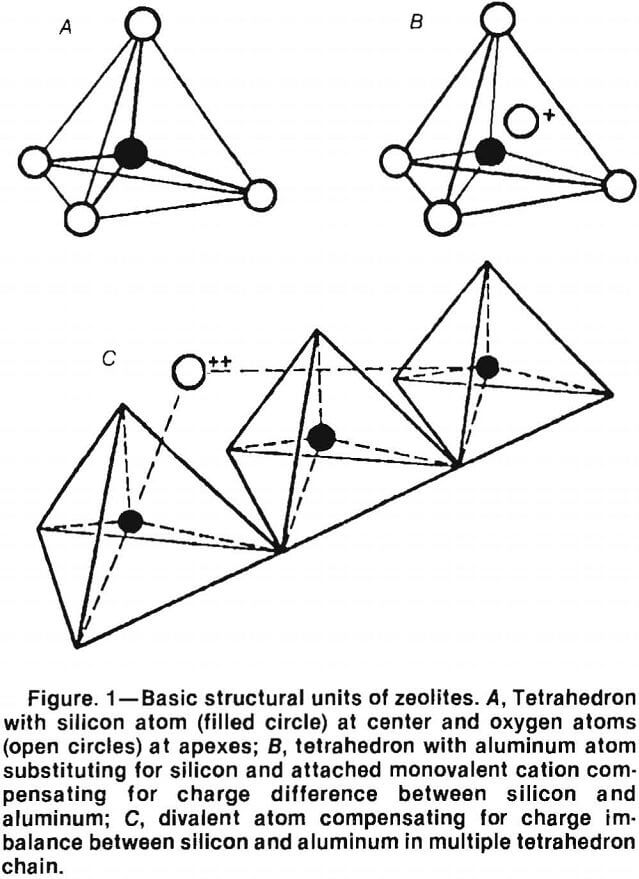
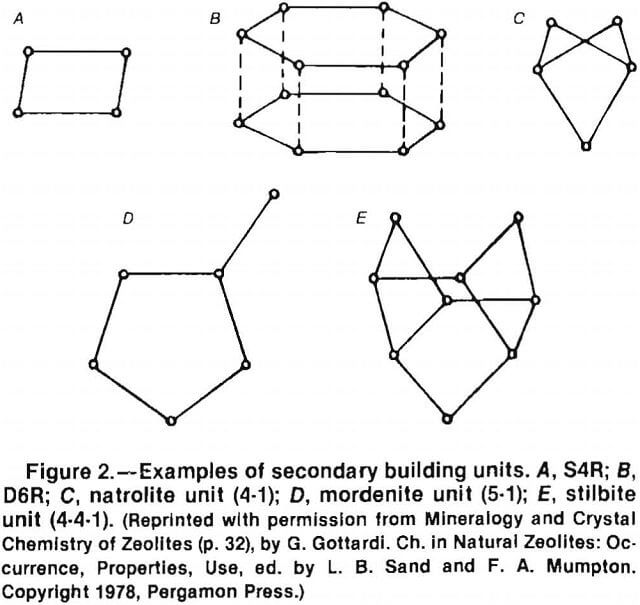
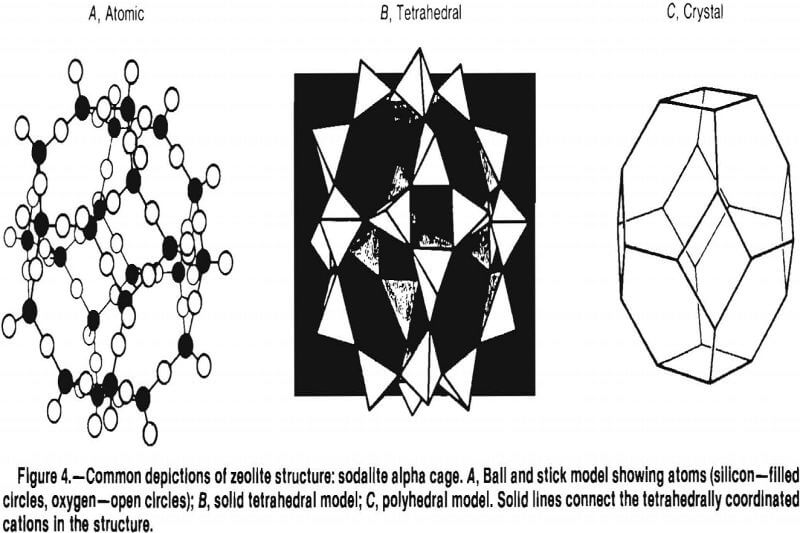
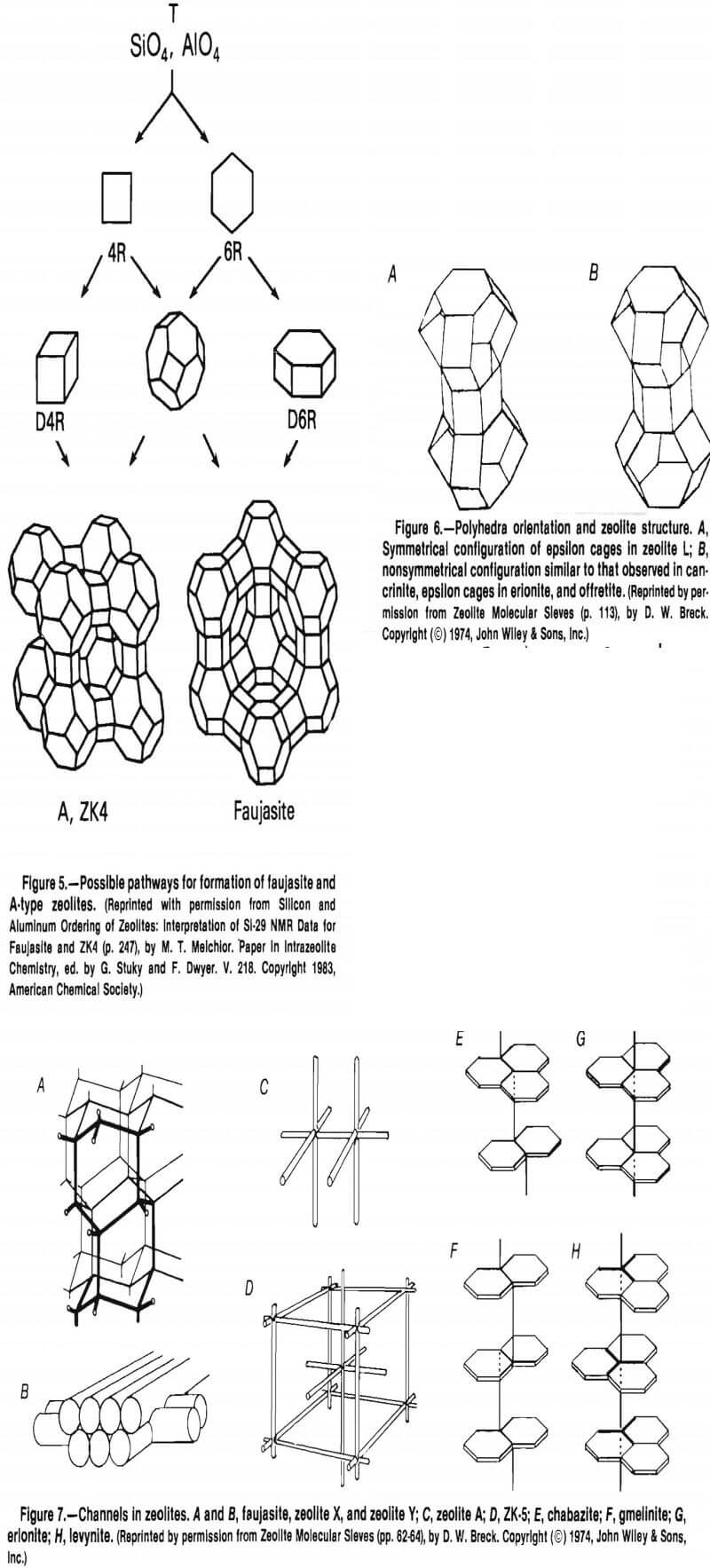
These tetrahedra are then grouped together to form secondary building units (SBU’s). There are eight SBU’s: the single 4-ring (S4R), the single 6-ring (S6R), the single 8-ring, the double 4-ring (D4R), the double 6-ring (D6R), the natrolite unit (4-1), the mordenite unit (5-1), and the stilbite unit (4-4-1) (fig. 2). Only the silicon atoms are illustrated, for clarity. The D4R and D6R units are combinations of the S4R and S6R units. The SBU’s can be assembled to form a variety of polyhedra (fig. 3). These polyhedra are normally depicted as the “ball and stick” model (fig. 4A), the “solid tetrahedral” model (fig. 4B), and the “polyhedral” model (fig. 4C).
The alpha cage or truncated cuboctahedron depicted in figure 4C is a component of more than one zeolite. Zeolite A is composed of alpha cages joined by D4R’s (fig. 5 ). If these cages were joined by D6R’s, faujasite or zeolite Y would be formed, with entirely different topology, cavity size, channel size and number, and different molecular sieving properties.
Different zeolites with distinctive characteristics and properties can also be produced using the same SBU (D6R) and the same polyhedra, by changing the orientation of the polyhedra with respect to each other (fig., 6).
Cavities and channels are created within the zeolite structure when the SBU’s are assembled. There can be as many as three channel types: nonintersecting, intersecting in two dimensions, or intersecting in three dimensions. In figure 7 from Beck, A and B illustrate the channel system in faujasite and zeolites X and Y: A shows the four connected networks, and B, the channels as an array of overlapping tubes. Figure 1C is a diagram of the three- dimensional system of the intracrystalline channels in zeolite A. Figure 7D is a diagram of the two independent sets of channels in paulingite. Both sets are of the same size but do not intersect. The rest of the panels in figure 7 are schematic illustrations of the main intracrystalline channels in zeolites that have frameworks based on parallel arrays of 6-rings: E, chabazite, F, gmelinite, G, erionite, and H, levynite.
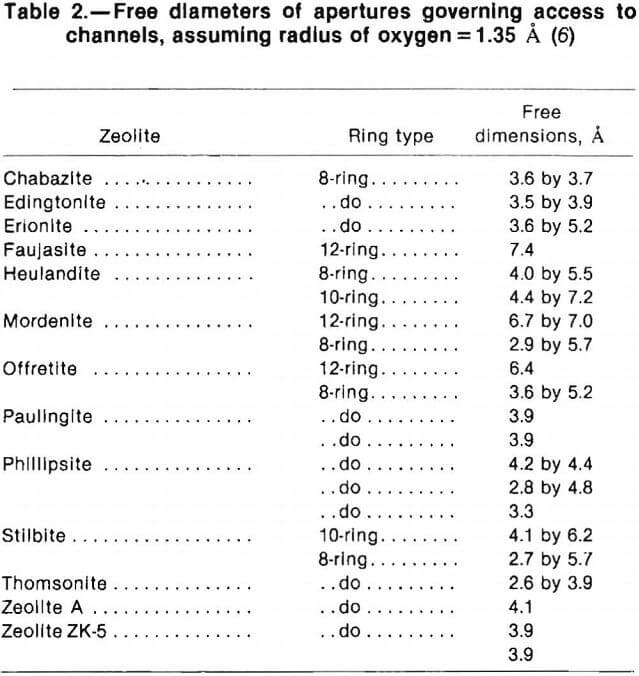
Channel sizes in a given zeolite can vary significantly depending on which cation is present, temperature and thermal vibration, and structural deformations. Theoretical calculations based on the number of tetrahedra forming a channel opening or aperture and assigning to oxygen ions the accepted 2.7- A diameter give a good approximation of the available channel size. Table 2, selected from Barrer, gives these calculated dimensions for ring sizes observed in zeolites. Six-ring apertures, present in many of the zeolites, and the channels limited by them are omitted because these rings allow entry only to the smallest polar molecules, such as water.
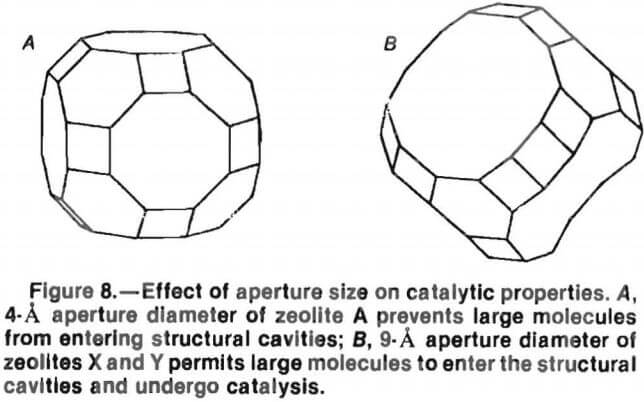
The cavities within the zeolite structure are accessed through these apertures in the polyhedral framework structure. Miller discussed the importance of aperture size, using the truncated octahedra of zeolite A, faujasite, and X and Y zeolites as examples. Since the apertures of zeolite A are too small to allow large molecules to enter the inner cavity, these crystals are limited in their range of catalysis.
However, the apertures can serve as molecular sieves for selective catalysis inside the cavities. Molecules that can enter the cavities and be catalyzed include oxygen, ammonia, hydrogen sulfide, methanol, sulfur dioxide, straight-chain hydrocarbons, and water.
Miller stated that because the apertures of the truncated octahedral polyhedral framework are large enough to permit virtually any molecule to enter the cavity, they do not function as molecular sieves. These large apertures, however, allow metal atoms to enter the cavity and substitute for alkaline and alkaline-earth metals in the structure. A wide range of molecules (including cyclopentane, benzene, fluorine compounds, high-molecular-weight hydrocarbons, alkyl aromatics) can enter the cavity to participate in reactions that take place on or near these catalytic metals (fig. 8).
The dimensions in table 2 refer to theoretically ideal, hydrated zeolites. Complete dehydration can produce irreversible structural changes. Stacking faults can block channels or reduce the effective aperture size. Entrained impurities can make large differences in effective channel sizes, and ion-exchange replacement of cations with those of different ionic radii change the effective channel size. A good example of this is that zeolite A is sold as “3A” (effective 3-A channel size), “4A,” or “5A.” The aluminosilicate framework topology is identical for all three, but different cations effectively change channel size.
Zeolite classification has evolved with increasing knowledge about the structure of zeolites. Earlier classification based on morphology had erionite, for example, grouped with other fibrous structures. Recent classifications are based on the framework topology. Breck grouped zeolites by common SBU’s into seven categories (table 3). No zeolite is typical of all other zeolites within a group because of slight differences in chemistry and crystallography.
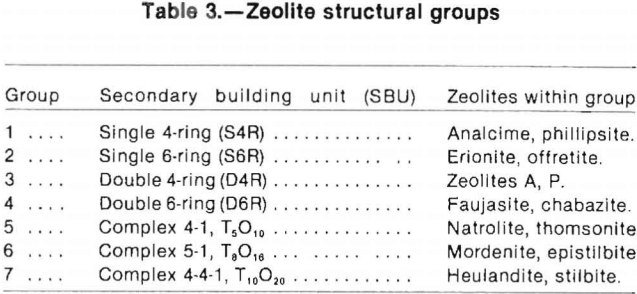
Formation Processes and Geologic Occurrence
According to Hay, most zeolite occurrences in nature are assignable to six types of geologic environments or hydrological systems: (1) saline, alkaline lakes, (2) saline, alkaline soils and land surfaces, (3) sea-floor sediments, (4) percolating water in an open hydrologic system, (5) hydrothermal alteration, and (6) burial diagenesis or metamorphism. Iijima lists two additional geological occurrences; magmatic zeolites and zeolites forming in impact craters. The largest and most commercially valuable deposits within the United States are those of categories 1, 2 and 4.
Zeolites commonly form by direct precipitation from pore fluids as in magmatic and some hydrothermal occurrences or by the alteration of volcanic glasses or poorly crystalline silicate minerals. The most common parent materials are volcanic glass, clays, montmorillonite, plagioclase, nepheline, biogenic silica, and quartz. Under the proper conditions, one zeolite species may replace another species. Temperature, pressure, chemical activity of ionic species, and the partial pressure of water all affect which species of zeolite forms. Temperatures of formation can range from ambient to 700° C, and pressures can range from 1 to 1,000 atm.
Figure 9 from Hay depicts the modes of occurrence of natural zeolites and the approximate depths at which these zones may be found.
Zone A in Figure 9 is characterized by nonanalcimic, alkali-rich zeolites, zone B by analcime or heulandite, and zone C by K-feldspar in 9 A, B, C, and D and by albite with or without laumontite in 9 E and F.
Saline Alkaline Lakes
Saline, alkaline lake environments are associated with two types of tectonic settings in arid and semiarid regions: block-faulted terrains and trough valleys associated with continental rifting. These settings establish a closed basin system and control the transport of clastic material beyond the basin edges. Both are essential for controlling lake chemistry.
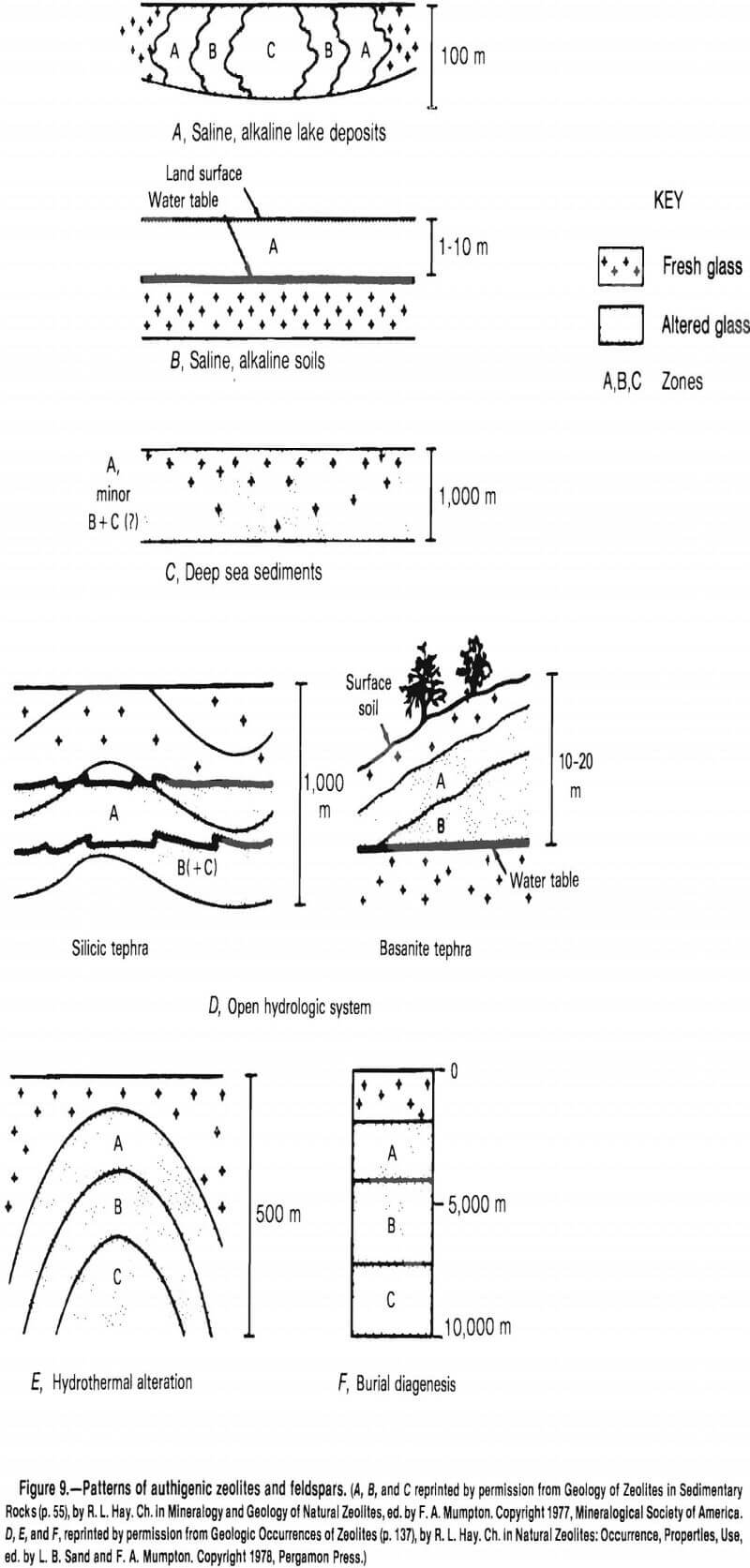
The restricted lakes in which zeolites form are alkaline, with pH values of 9.5. Analcime, clinoptilolite, phillipsite, erionite, chabazite, and mordenite commonly form, replacing volcanic glass, biogenic silica, poorly crystallized clay, montmorillonite, plagioclase, nepheline, and quartz. These deposits are zoned laterally, as in figure 9 A, representing the changing chemistry of the water as evaporation and precipitation of minerals proceed.
The zonation of the mineral beds in the various saline, alkaline lakes indicates the sequence of the zeolite diagenesis.
- Glass → alkalic, silicic zeolites.
- Alkalic, silicic zeolites → analcime.
- Analcime → K-feldspar.
- Alkalic, silicic zeolites → K-feldspar.
Some controlling chemical parameters during the glass-to-zeolite reactions are the cation ratios, silicon-aluminum ratios, and the activity of the water. Dissolution of the glass is controlled by the salinity and pH. These reactions can be rapid, with vitreous tuffs altering to zeolites in less than 1,000 yr. Ancient Lake Tecopa, shown in figure 10, is typical of the saline, alkaline lake environment.
Saline Alkaline Soils
Climate is the controlling factor in the formation of zeolites in saline, alkaline soils. These deposits form in arid and semiarid regions where evaporation causes sodium carbonate-bicarbonate to concentrate in the surface soils. Rainwater percolating through the soils dissolves the sodium carbonate-bicarbonate and increases in pH, allowing it to alter glasses and aluminosilicates present in the soil. The water table is the probable limit of zeolitic alteration processes. Zeolite deposits of this type can approach 18 m in thickness and contain 15% to 40% zeolites. Analcime is the most common zeolite forming in these formations. Minor amounts of phillipsite, natrolite, and chabazite also occur. Deposits of this nature have been observed in the Olduvai Gorge, Tanzania, and other sites in Africa and in the Western United States.
Marine Sediments
Zeolites form in marine sediments under low temperatures and moderate pH conditions (pH values of 7 to 8). Zeolites in the slow sedimentation regions of the Pacific and Indian Oceans occur in post-Miocene brown clays, vitric siliceous and calcareous oozes, and basaltic volcanic sediments. Phillipsite is the dominant species. Those in the rapid sedimentation regions of the Atlantic and Pacific margin are in calcareous sediments and terrigenous clays of Paleogene and Cretaceous ages. Clinoptilolite is the dominant species here. Analcime, chabazite, erionite, laumontite, gmelinite, natrolite, and thomsonite also may be present in these marine deposits.
Zeolites in sea-floor sediments form by the reaction of glasses with pore water. Except for the silica content, which ranges from 4 to 65 ppm, sediment pore water is similar to seawater in composition. Phillipsite is associated with the low silica concentrations (less than 20 ppm) typical of basaltic tephras, and clinoptilolite is associated with the high silica concentrations (20-40 ppm) of siliceous tephras. Clinoptilolite occurring with phillip-site probably forms when basaltic glass reacts with silica- rich pore waters.
Boles and Wise suggest that since marine clinoptilolite occurs as euhedral crystals, their formation must have been from pore water rather than as a direct replacement of earlier phases. In spite of this, they think that because phillipsite is found in the younger sediments it is likely that the clinoptilolite in the older sediments replaced it and that time was an important factor.
Open Hydrologic Systems
Zeolites in open systems form from the percolation of ground water through porous pyroclastic materials rich in reactive glass. The pH and dissolved solid content of the ground water increase as it reacts with the vitric ash, until zeolites are precipitated. Movement of the ground water downward through the system results in a vertical zonation of water composition and authigenic minerals, including zeolites. Typically a silicic tephra may contain an upper zone of fresh glass, montmorillonite, and opal. The next bed can contain up to 90% clinoptilolite, and the underlying zone may contain analcime, potassium, feldspar, and quartz. Alteration of the glass can occur rapidly because of the high pH (approximately 9.5) resulting from hydrolysis.
An example of an open-system deposit is the basanite vitric tuffs of Koko Crater in Hawaii. These tuffs consist of 1.5 to 12 m of fresh glass, opal, and montmorillonite, The next lower zone consists of 1.5 to 10 m of palagonite tuffs with phillipsite and chabazite. The lowest zone is 8 m thick and predominantly analcime.
Hydrothermal Systems
Zeolites precipitate in hydrothermal systems from alkaline to weakly acidic hot water. The assemblages observed are controlled by temperature, host rock composition, host rock permeability, and geothermal fluid composition. Clinoptilolite and mordenite occur in the shallowest and coolest zones. Analcime or heulandite and laumontite or wairakite, the less hydrated forms, occur in the deeper and hotter zones. Submarine hydrothermal activity may be responsible for the formation of some zeolites in deep-sea sediments.
Examples of hydrothermal zeolite deposits are found in Yellowstone Park, WY; Wairakei, New Zealand; and Onikobi, Japan. At the Onikobi site in Japan, there are four zones: a mordenite zone, a laumontite zone, a laumontite-wairakite zone, and a wairakite zone.
Burial Diagenetic Systems
Zeolites associated with burial diagenesis occur in thick volcanoclastic sediments that were metamorphosed at increased temperatures. Two reaction sequences are recognized, the alkali zeolite reaction series and the calcic zeolite reaction series. The alkali series includes the zeolites clinoptilolite, mordenite, and analcime. The calcic series includes clinoptilolite, heulandite, and laumonitite. Burial diagenetic sequences are typified by vertical zonation. This mineralogical zonation is related to increasing burial depth. Iijima recognized five zones based on the mineralogy of marine sediments subjected to burial diagenesis (fig, 11). Temperatures increase with depth, as in the Green Tuff region in Japan where temperatures were 41° to 49° C at the top of the zeolite sequence and 120° to 124° C at the base of the sequence. More hydrated zeolites occur at shallower depths. The upper zones of burial diagenetic sequences are mineralogically similar to open hydrologic systems. The major distinction is the gradual change between zones in the burial diagenetic sequence, compared with the sharp contacts of the open system deposit.
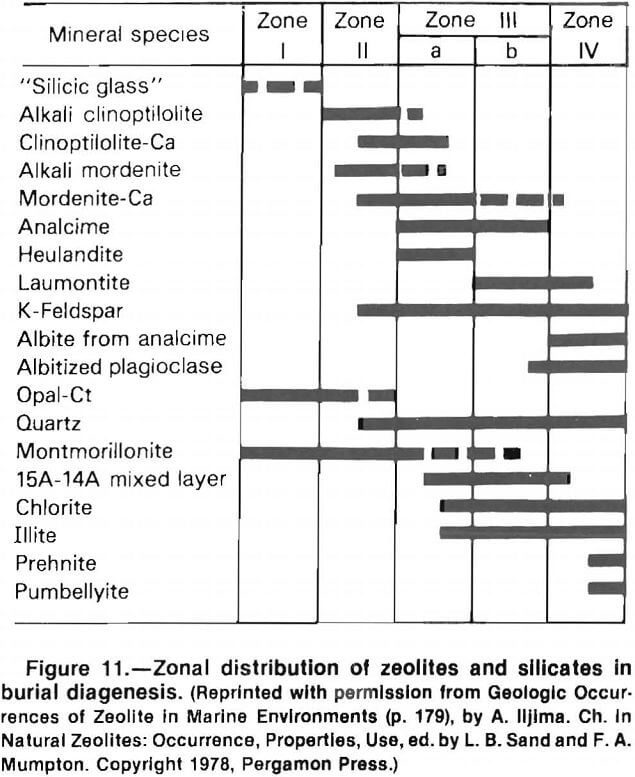
An example of burial diagenetic deposits is the Green Tuff region of Japan. In the Niigata oilfield, the upper zone is 0.8 to 1.9 km thick, containing fresh glass. The next zone, 1.6 to 2.5 km thick, contains mordenite and clinoptilolite. This is followed by an analcimic zone, 1 km thick, and an albitic zone, 0.7 km thick. Analcime is pseudomorphic after mordenite and clinoptilolite. Temperatures range from 41° to 124° C.
Magmatic Systems
Zeolites crystallize during the late stages of formation of magmatic rocks. They commonly occur as fine crystals lining vugs in basic igneous rocks, although they also occur as interstitial grains and globules. Crystalline zeolites form through interaction of fluids with the surrounding rock. Zeolite globules form when water-rich magma separates into two immiscible liquids.
The zeolites that form under magmatic conditions are grouped by bulk composition. Aluminum-rich zeolites occur in basic igneous rocks with low silicon-to-aluminum ratios. Aluminum-poor zeolites occur in igneous rocks richer in silicon. Several zeolites that occur in igenous rocks are analcime, clinoptilolite, heulandite, mesolite, mordenite, natrolite, phillipsite, and stilbite.
Impact Craters
Iijima reports zeolites occurring in the Nordlinger Ries impact crater in West Germany. In this occurrence, glass formed by the meteor impact altered to analcime and smectite. Microcavities in the deposits were filled by analcime, clinoptilolite, erionite, harmotone, and phillipsite.
Exploration
Extensive exploration for natural zeolite deposits did not begin until the 1950’s, when Union Carbide Corp., through its Linde Division, initiated a search for minable quantities of natural zeolites that might compete with their synthetic zeolites (SI). In describing the onset of this project, Mumpton reports that mineralogical and mineral- commodity consultants employed by Union Carbide did not believe that minable deposits of zeolites would be found. They, like most researchers, believed that zeolites were mineralogical curiosities that filled vugs and fractures in igneous rocks. However, in the 5-yr effort, over 3,000 samples were collected and approximately 200 new zeolite occurrences in volcano clastic sedimentary rocks were discovered.
As the initial search was going on, a thorough literature search revealed earlier discoveries of sedimentary deposits. Coombs, at the University of Ofago in New Zealand, described the widespread occurrence of laumontite, analcime, and heulandite in burial metamorphic sequences of pyroclastic sediments in that country and demonstrated the occurrence of substantial amounts of zeolites in “nonigneous” environments. Ames, Sands, and Goldich also described large occurrences of clinoptilolite at Hector, CA. This information prompted Union Carbide’s Linde Division to intensify its investigation of zeolite occurrences in pyroclastic rocks.
Samples from the Hector deposit were examined by standard X-ray diffraction and microscopic techniques (SI). Mumpton reported that “several fine-grained, homogeneous, lightweight samples from the Hector deposit, representing beds as thick as 12-15 ft. were found to contain as much as 80-85% clinoptilolite, and that the Hector samples demonstrated conclusively that near-monomineralic deposits of natural zeolites could indeed be found in mineable quantities within the continental United States. They also demonstrated that the few reports in the earlier geological literature about the occurrence of zeolites in sedimentary rocks of volcanic origin were not flukes, but that many more such deposits were likely to be found in geologically similar environments throughout the western part of the country and probably throughout the world.
Early in 1958, a sample described as erionite from a million-short-ton, easily mined, sedimentary deposit was submitted to Linde. Laboratory examination indicated that the sample contained 85% to 90% erionite and that its chemical, adsorption, optical, and X-ray diffraction properties closely matched those of Linde’s synthetic analog, zeolite T. Later, the Kennedy Minerals Co., discoverer of the deposit, revealed that the deposit was located in a broad basin of lacustrine and fluviatile sediments about 2 miles west of Rome, OR. Here, the Owyhee River had cut through plateau basalts to expose several hundred feet of volcanogenic sedimentary rocks.
The Linde exploration program remained active until 1961. As it tapered off, Shell Development Co. started a modest exploratory effort led by H. D. Curry. Regnier, at Columbia University, described beds of high-grade erionite, clinoptilolite, and an unidentified zeolite (later identified as phillipsite) in altered Cenozoic strata in Pine Valley, NV. Deffeyes reported that these same zeolites had been recognized in near-monomineralic beds of altered tuff in Jersey and Reese River Valleys, NV, and that zeolites seemed to be common in the rocks of the Rocky Mountain west. Zeolites had not been previously recognized in these rocks because of their extremely fine-grained nature (1 to 5 µm) and the tendency of the field geologist and stratigrapher to limit their laboratory studies of these materials to microscopic examinations. Once X-ray diffraction techniques were employed, the presence of zeolites became much more widely recognized in Cenozoic volcanogenic sediments.
Table 4 from Mumpton lists the discovery dates of 32 prominent natural zeolite deposits. Nearly 90% were discovered between 1957 and 1962.
Mumpton’s own summary of his paper is given in its entirety below because it contains so much of the story of natural zeolites in the United States:
In the five-year period beginning in January 1958, and ending in December 1962, more than 3000 samples were collected and approximately 200 new occurrences of zeolite minerals in volcanoclastic sedimentary rocks were discovered as a result of the exploration efforts of Union Carbide Corp. Nearly 100 other occurrences of these minerals in the United States became known at the time as a result of other discoveries by university geologists (e.g., L. B. Sand, F. B. Van Houten, R. L. Hay, K. S. Deffeyes, Jerome Regnier) and by employees of the U.S. Geological Survey (e.g., R. L. Smith, R. E. Wilcox, H. A. Tourtelot, TheodoreBotinelly, A. D. Weeks, D. L. Peck, R. C. Erd), and from the literature searchings of these individuals and Linde’s research personnel.
Much of Carbide’s exploration effort was guided empirically by the newly developed knowledge that zeolites were abundant in (1) flat-lying, light colored tuffaceous beds of Cenozoic age which commonly crop out as erosion resistant ledge-formers in association with unaltered volcanic ash, bentonites, and other typical lacustrine formations; and (2) light-colored, altered ash-flow tuffs in Tertiary pyroclastic bodies commonly associated with perlites and welded tuffs.
Aerial reconnaissance of Tertiary basins, the field test developed by Linde, and the routine use of X-ray diffraction for the identification of exploration samples proved to be effective. The direct communication between the field geologists and the chemists and mineralogist at Linde and the short turnaround time for field samples gave rise to technical assistance that could not be provided by the Nuclear Division, which itself had no previous experience with zeolites or with non-metallic commodities in general.

Where in 1957, zeolites were thought to exist primarily as late-stage hydrothermal products in amygdaloidal basalts, by 1962 they were being recognized as common constituents of altered pyroclastic material in a wide variety of sedimentary and volcanic environments. During this period, the heretofore rare zeolite erionite was found to occur in large, mineable deposits, and the widespread distribution of clinoptilolite in such rocks was firmly established. The first verified occurrences of chabazite and mordenite in sedimentary rocks of the United States were also discovered during the Carbide program. The flat-lying, near-surface, and near-monomineralic nature of the deposits suggested that many would eventually be mined and that the so-called sedimentary zeolites would one day become an accepted industrial mineral commodity.
The sudden interest in zeolites was further emphasized by the fact that of the more than 400 published reports that describe zeolites in sedimentary rocks throughout the world, more than 75% were published in the 1950’s and 1960’s.
Synthesis
The synthesis of zeolites was first reported by St. Clair Deville in 1862. By heating aqueous solutions of potassium silicate and sodium aluminate in a glass tube at 170° C, Deville produced the zeolite levynite. In 1882, De Schulten reported the synthesis of analcime. However, data are not available to substantiate these and other experiments, and much of the early work cannot be duplicated in the laboratory. The first substantiated synthesis of zeolites was not performed until the 1940’s when X-ray diffraction was used to identify phases. Prior to this time, light microscopy was used for phase identification, and the fine-grained nature of synthesized zeolites made identification difficult. A list of zeolites synthesized prior to 1936 is presented in table 5.
Early attempts at zeolite synthesis sought to duplicate the conditions under which zeolites were thought to crystallize in basaltic rocks. In 1959, R. M. Milton and associates at Union Carbide Corp. suggested a new approach, which allowed the synthesis of zeolites at low temperatures. Their method used highly reactive ingredients in a closed system at low temperatures, often below the boiling point of water. This made the large-scale production of synthetic zeolites feasible.
Breck reports the general conditions used in zeolite synthesis:
- Reactive starting materials such as freshly co-precipitated gels or amorphous solids.
- Relatively high pH introduced in the form of an alkali metal hydroxide or other strong base.
- Low-temperature hydrothermal conditions with concurrent low autogeneous pressure at saturated water vapor pressure.
- A high degree of supersaturation of the components of the gel, leading to the nucleation of a large number of crystals.
The following are simultaneous reactions that occur during the synthesis process :
Precipitation of a gel phase,
Dissolution of the gel,
Nucleation of zeolite(s),
Continued crystallization and crystal growth of the zeolite(s),
Dissolution of the initial metastable phase(s),
Nucleation of a more “stable” metastable phase or phases,
Continued crystallization and crystal growth of the new crystalline phase(s) while the initial crystals are dissolving, Dissolution of the metastable phase(s),
Nucleation of the equilibrium phase(s),
Crystallization and crystal growth of the final crystalline phases.

Because of the multitude of reactions, time is an important factor determining phase stability and crystal ordering. Breck states “that a mineral zeolite which has existed over long periods of geological time and a synthetic zeolite with a related structure but which was synthesized rapidly in the laboratory may exhibit differences in properties due to the ordering that may occur in the mineral as opposed to the lack of ordering the synthetic structure shows.” Many of the synthetic zeolites that are not structurally related to any natural zeolite may be nonequilibrium phases. Metastable synthetic structures may also explain why many natural zeolites do not have synthetic counterparts despite the synthesis of over 100 different zeolite types.
Several processes are used to synthesize zeolites. In one process, kaolin is used as the source of the silica and alumina required in the reaction because of its higher reactivity. Equations 1 and 2 (modified to omit unreacted water) give the reaction sequence.
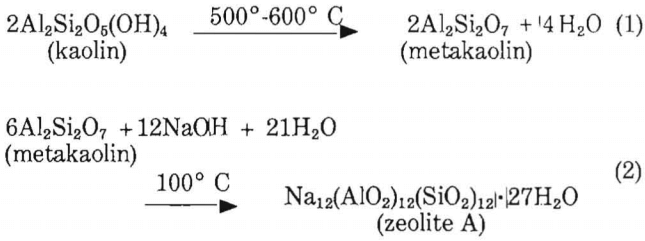
A material balance of equation 2 shows that for each short ton of zeolite A produced 1,217 lb of metakaolin are needed, 438 lb of sodium hydroxide, and 345 lb of reaction water. Other water is needed to provide the necessary aqueous environment but does not react. For equation 1, 1,414 lb of kaolin are necessary to produce 1,217 lb of metakaolin.
Zeolites can also be synthesized from aluminosilicate gels of the alkali earths, either potassium, sodium, lithium, rubidium, and/or cesium. In this process, the starting gel is depolymerized by the hydroxyl ions present in the starting mixture, and zeolite crystallites form. Magee and Blazek discuss the synthesis of zeolite Y using silica gels as a basic ingredient. The procedure they report is as follows:
- Place 2,090 g H2O in a resin kettle.
- Dissolve 162.2 g NaOH in the water.
- Add 425 g of sodium aluminate, containing 24 wt % Al2O3, 20 wt % Na2O, and 56 wt % H2O.
- Cool to 100° F, and then add 570 g of calcined silica.
- Cold-age at 100° F for 24 h with mild agitation.
- Increase temperature to 212° F, and age for 24 h with mild agitation.
- Quench with water, and filter to remove the solids from the mother liquor.
- Wash with hot water, and dry in forced-air oven at 250° F.
This process yields 464 g of sodium-Y zeolite (Na2O·Al2O3,5SiO2·nH2O). The silicon-aluminum ratio is 2.5 in this process; it was 1 for zeolite A. Small differences in molar ratios can produce entirely different zeolites under identical conditions. The form of the starting materials can also affect mineral stability. Type A, X, and Y synthetic zeolites are examples of synthesis from a gel. Type A as synthesized or as modified, was the first zeolite to become available in sufficient quantities to permit extensive laboratory study. Types X and Y had sufficient pore size, cage volume, and accessibility to selectively permit entry and controlled reaction of the larger molecules common to the petrochemical industry.
The main difference between the X and Y catalysts is the silicon-aluminum ratio. The silicon-aluminum ratio for zeolite X ranges from 1 to 1.5 while that for zeolite Y ranges from 1.5 to 3. The diameter of the large cage of both is 13 A, but the unit cell of X is slightly larger, 25.02 to 24.86 A, while that of Y is 24.85 to 24.61 A. Table 6 lists some of the zeolites that can be synthesized from the Na2O·Al2O3·SiO2·H2O system and the reactants used. Most of these zeolites have a sodium oxide-to-alumina ratio of 1.
Quaternary ammonium hydroxides, such as tetramethyl ammonium ((CH3)4·NOH), can also be used as a source of cations in zeolite synthesis. The ammonium compounds act as templates to control the structure of the zeolite. This process led to the synthesis of some new and technically important zeolites such as Mobil’s ZSM-5. The basic process consists of reacting an aluminosilicate gel containing sodium and tetrapropyl ammonium cations. Varying silica-alumina ratios are obtained by adjusting the silica-to-alumina ratio in the starting reaction mixture. Such adjustments do not alter the framework topology.
The recurring arrangement of tetraheda into five-membered rings is evident in figure 112, which shows both a one-dimensional and a two-dimensional view of the structure of ZSM-5.
The channel system of ZSM-5 consists of a 3D intersection of a straight channel with elliptical cross-section .51 x .57nm along the orthorhombic b-axis (1.98nm) and a zig-zag channel along [101] and [107] directions (a = 2.01; c = 1.34nm). Four channels meet at each intersection (fig. 13). To avoid possible confusion caused by the perspective, the dimensions of the channels along the b-axis are
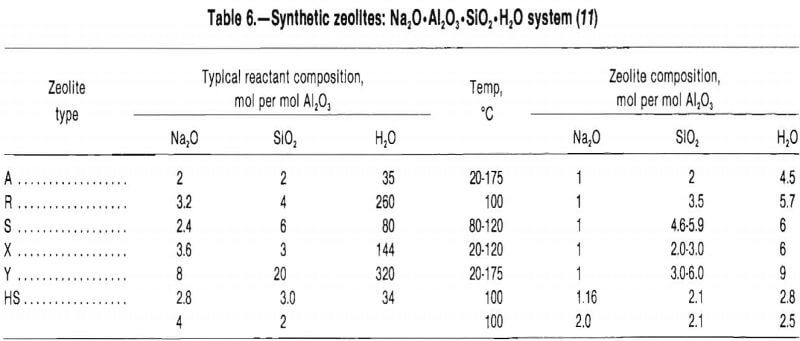
Note.— Reactants for zeolites A, R, S, X, and Y are sodium aluminate, sodium silicate, sodium hydroxide, and colloidal silica; reactant for zeolite HS is silica gel.
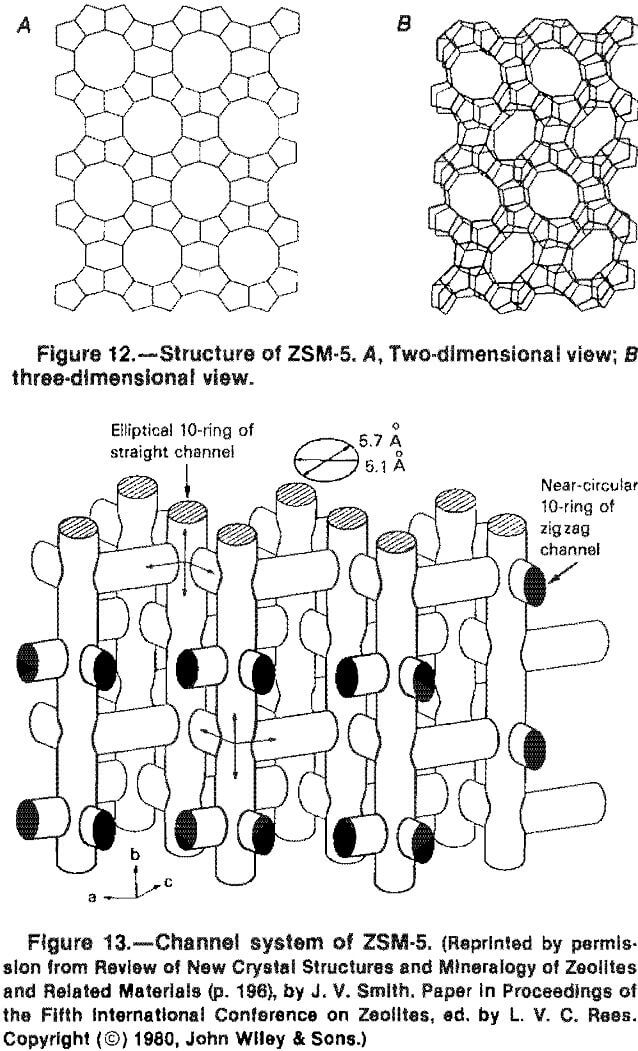
shown at the top of the figure. The straight channels along the b-axis are seen from above in figure 12.
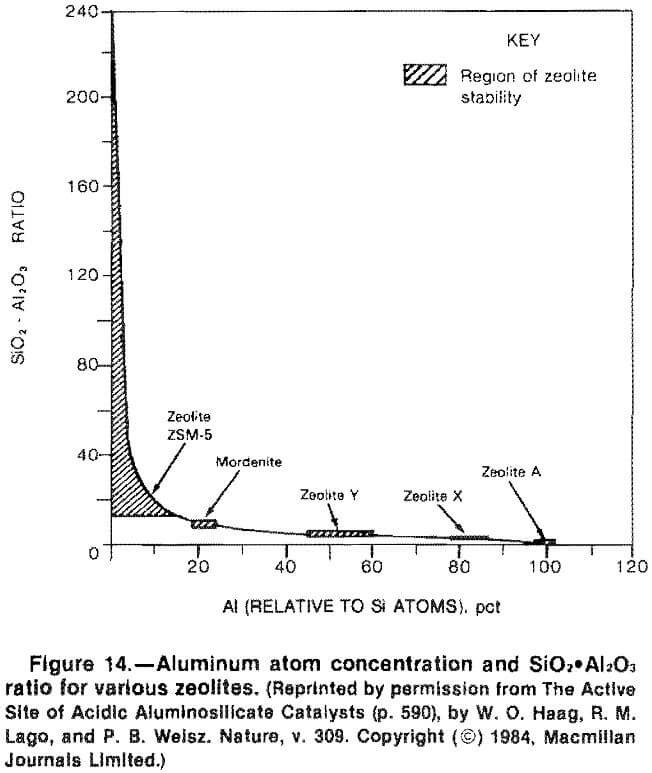
The synthetic zeolite ZSM-5 has an almost infinitely variable silica-alumina ratio (fig. 14) Smith reports the
formula of the sodium form as NanAlnSi96-nO192·16H2O, with n less than 27 but typically equal to 3. Whereas a structure with tetrahedral Al and corresponding exchange sites shows ion-exchange and reversible dehydration expected of an ideal zeolite, the Al-free structure does not show ion-exchange and is hydrophobic and organophilic: thus silicalite is a molecular sieve but not a zeolite.
Aluminum atoms are no longer structure-determining constituents; that is, they no longer contribute a major portion of the framework structure. The average distance between aluminum sites is equivalent to several atomic diameters, and their distribution should approach randomness.
Chemical and Physical Modification
Dehydration and Rehydration
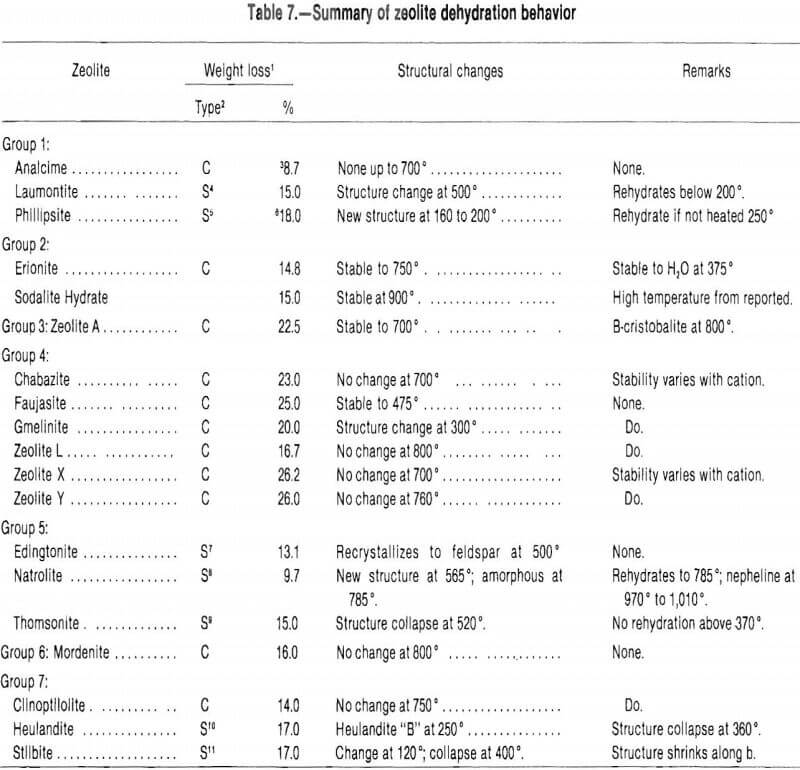
The dehydration properties of zeolites are important because they influence the adsorption characteristics of the zeolite. Dehydration can cause distortion of crystalline structure. This influences the volume and shape of the micropores. Zeolites that retain structural stability during dehydration have micropores that fill and empty reversibly so that adsorption is a matter of pore filling, and the surface area concepts and calculations common to other solid absorbents do not apply. Dehydration can also affect the charge distribution within the zeolite structure. This occurs when cations coordinated with water migrate to different crystallographic sites upon dehydration. Table 7 summarizes the effects of dehydration on some zeolites. A wide variety of temperatures at which structural changes occur and an even greater variety of temperatures at which there are steps in the weight-loss curves of zeolites are demonstrated by thermogravimetric analysis.
Structural Hydroxyl Groups
Structural hydroxyl groups within the zeolite structure are desirable for catalytic processes because they provide active sites for hydrocarbon conversions and other catalytic reactions. Hydroxyl groups can be introduced by two techniques, cation hydrolysis and deammoniation of NH4- exchanged zeolites. Cation hydrolysis can be induced by normal dehydration, where a water molecule is dissociated, the hydroxyl is lost, and the hydrogen ion is fixed to structural oxygen atoms. Treatment of the zeolite with a strong acid
will also produce hydrolysis through hydrogen exchange with available cations. Acid treatment, however, can cause structural degradation, especially in aluminum-rich zeolites. Deammoniation can also be used to create structural hydroxyl groups (H). The ammonium ion is thermally decomposed, ammonia is evolved, and the hydrogen ion is fixed in the structure. By controlling the amount of deammoniation occurring, the number of structural hydroxyl groups formed can be regulated.
Sorption and Diffusion
Molecular Sieving
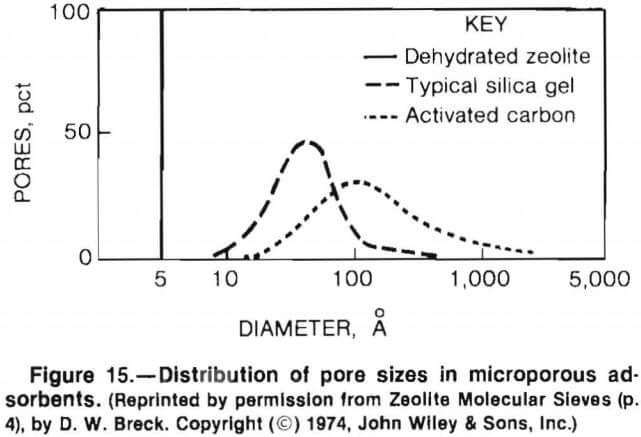
Molecular sieving refers to the selective adsorption of cations within a sorbent, based on physical dimensions and charge distribution. Molecular sieving depends on the characteristics of both the sorbent and the material being adsorbed. Zeolites are well suited for molecular sieving because they possess an open, yet uniform crystalline framework that has a narrow pore size distribution. This contrasts with other types of molecular sieves, such as silica gels or activated carbon, which have a wider range of pore size distributions (fig. 15). This property makes zeolites more size selective than other molecular sieves.
Molecular sieving can be affected by heating and dehydration of the zeolite. Heating results in distortion of the crystalline framework and in aperture enlargement due to the increased vibrational amplitude of the structural oxygen as temperature increases. Dehydration results
in cation displacement and subsequent changes in the charge distribution within the zeolite structure. Dehydrated zeolites selectively adsorb polar molecules, such as H2O, CO2, and H2S. This selectivity results from the unusual charge distributions within the structure due to the positions of cations, hydroxyls, and the substitution of aluminum for silicon in the framework structure.
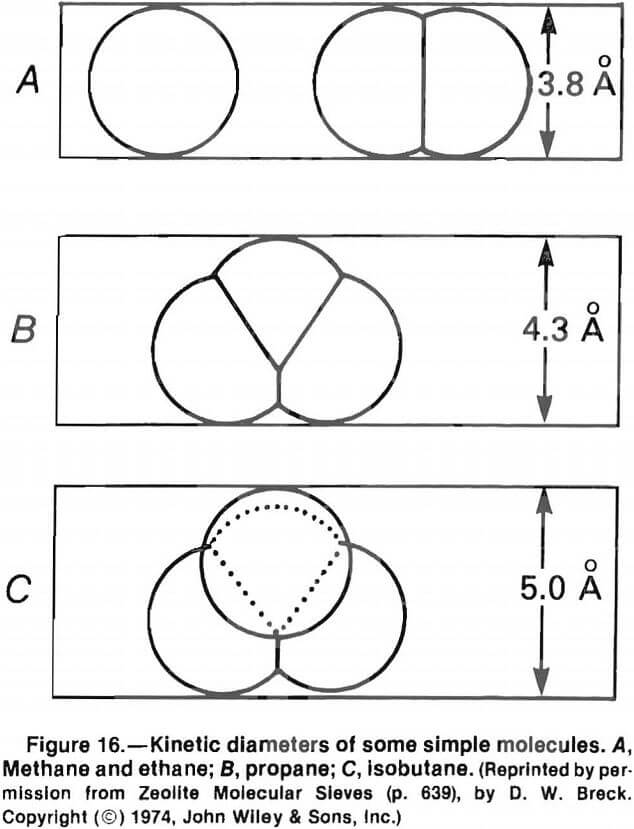
The size, shape, and charge of the adsorbed phase also influence molecular sieving. For instance, rubidium with a radius of 1.48 A is extensively exchanged by analcime, while sodium with a radius of 1.65 A is not exchanged. Similar effects are observed with organics because of differences in their sizes and shapes (fig. 16).
Polar molecules with their uneven charge distributions are selectively adsorbed by some zeolites.
Diffusion
Diffusion is the migration of sorbate within the crystal. It affects selective adsorption, desiccation, molecular sieving, and catalysis. Diffusion in zeolites is very complicated. Diffusivity, which is affected by molecule size and shape and framework topology, has proven to be extremely difficult to predict. Maurer notes that with broader knowledge of zeolite diffusion processes, it has become clear that currently no general model for diffusivity prediction exists. Review of the available literature suggests that these data must, for the foreseeable future, be obtained empirically. Barrer lists the variables that influence these rates:
- Intracrystalline channel geometry and dimensions.
- Shape, size, and polarity of the diffusing molecules.
- Cation distributions, size, charge, and number.
- Lattice defects such as stacking faults.
- Presence of impurity molecules in the diffusion pathways.
- Structural changes brought about by penetrants.
- Structural changes associated with physical and chemical treatments.
- Concentration of diffusant within the crystals.
Barrer lists methods by which direct measurements of mass transport into or out of porous crystals can be obtained:
- The change with time of the volume of gas or vapor around the sorbent, keeping the pressure constant.
- The change with time of the pressure of gas or vapor around the sorbent, keeping the volume constant.
- The change with time in the weight of sorbent bathed in a constant pressure of sorbate vapor.
- The volume change with time of a liquid sorbate in a sorption pipette connected to the sorbent via the vapor phase.
- The transfer with time of tracer between gas or liquid phase and intracrystalline sorbed phase.
The mobilities or diffusion coefficients are inferred by examining properties related to sorbate content or mobility. These include
- Birefringence changes in single crystal plates that parallel changes of sorbate content.
- Sorbate mobility inferred from jump times derived from nuclear magnetic resonance studies.
- Sorbate mobility inferred from dielectric relaxation processes.
- Molecular motion evaluated by neutron-scattering spectroscopy.
- Changes with time, as the amount sorbed changes, in the intensity of specific infrared absorptions characteristic of the sorbate interacting with the sorbent.
Pore Volume
The availability of large volumes of internal space is one of the most desirable characteristics of zeolites. The amount available depends on so many variables that these data must be derived empirically. Some of these variables are
- Micropore accessibility-the size and geometry of controlling apertures, and any blockage of these by guest ions or stacking faults, can admit or occlude prospective sorbates.
- The size, shape, number, and location of cations.
- The size and shape of the sorbate molecules.
- Temperature.
- Pressure.
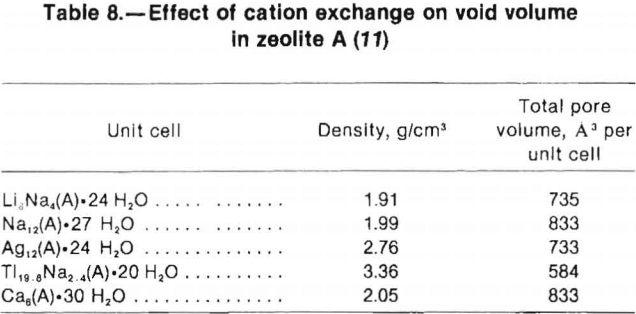
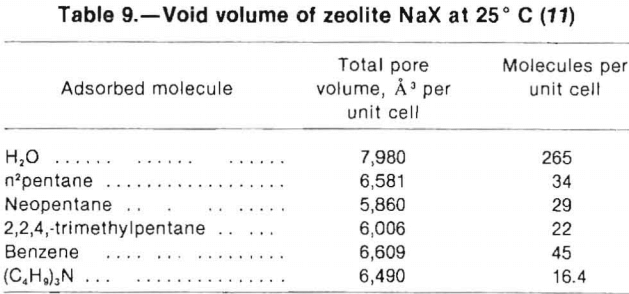
Table 8, from Breck, illustrates the effects of exchangeable cations on the available pore volume of zeolite A. Smaller void volumes are associated with the presence of large cations. For example, when large Tl cations replace Na cations the void volume is reduced by 30%.
These volume measurements were determined from the water contained in a fully hydrated zeolite. They give good comparative results, but if the zeolite structure contains cages inaccessible to molecules larger than water, then
the effective volume is reduced. Of the calculated void volume of zeolite A, for example, 16% is unavailable for molecules larger than water. Table 9, also from Breck, indicates the effect of different-sized sorbate molecules on void volume determinations. For larger or branched molecules, less void space is accessible and fewer molecules can be adsorbed per unit cell.
Zeolites and Catalysis
Sleight defined “a catalyst as a substance that accelerates a chemical reaction without itself being consumed. The degree of acceleration can be enormous, sometimes more than a factor of 10 10.” Many natural and synthetic zeolites are catalytic agents; many gain additional catalytic properties by modification and/or by acting as supports for other catalytic agents; and some catalyst mixtures are only partially composed of zeolites so as to gain both physical and chemical properties from the matrix material, which can also act as a dilutent to reduce overly high catalytic activity.
Sleight said that “one of the most useful zeolite structures is that of the naturally occurring zeolite faujasite.” Because natural faujasite is not readily available, synthesis is the primary source. The structure of faujasite is shown in figure 6. The silica-to-alumina ratio ranges from 1 to 3. Zeolites synthesized with this structure are generally referred to as either zeolite X (small silicon-aluminum ratio) or zeolite Y (large silicon-aluminum ratio). The free pore openings of the faujasite structure are the largest among zeolites, about 7.4 A.
Plank reported that the invention of zeolite cracking catalysts was in response to a need for a stable, selective catalyst of high activity. He looked for a catalyst whose active sites were located in controlled pores not much larger than the molecules to be cracked. Both molecular sieve zeolites and silica-alumina gels were considered, but stream regeneration significantly lowered the activity of the gel formulation while not affecting zeolite.
As pure zeolites had unacceptably high activities and needed diluting with a matrix that would add stability, the first experimental catalyst contained 25% sodium faujasite (Na X) base-exchanged with ammonium chloride in a silica- gel matrix. It gave desirable selectivity and activity and continued to function well at coke levels that would have deactivated previously used catalysts. Using ion-exchange mechanisms to introduce either calcium, manganese, or rare-earth active sites in the zeolite matrix, Plank produced catalysts that had at least 100 times the activity of the conventional silica-alumina catalysts.
Plank and coworkers developed several catalysts during the 1960’s that virtually revolutionized the refining of oil to produce gasoline and other products. Plank reported that, compared with conventional methods, the calcium catalyst produced 20% more gasoline; the manganese, 24% more; and the rare earth, 16% more. Coke decreased by 28%, 56%, and 40% respectively. Plank calculated that a 7% increment in gasoline could be expected in the United States, with an added product value of $140 million (1967 dollars) and a potential savings of 190 million bbl of crude oil years at the 1963 demand rate. These figures have increased in recent years with the major changes in the oil market. Magee and Blazek reported that, in the decade between the midsixties and the midseventies, between 10,500 and 15,000 st/yr of synthetic faujasite alone was used for the production of catalytic cracking catalysts. Clifton estimated that 92% of the world market for cracking catalysts was held by the zeolitic catalysts.
Maugh, in his study of the state of the art of catalysis, reported that the shape-selective systems were one of the most intensively studied of the new classes of catalysts. He said that they were developed to remedy one of the principal drawbacks of other heterogeneous systems: lack of selectivity. In some industrial reactions, a wide spectrum of products is generated. But for applications such as the production of high-octane gasoline, a narrow product range is desirable.
Zeolites are successful because of the size of their surface pores and interior cavities, where reactions take place. The pore size determines which molecules can enter the cavities to undergo catalysis and which molecules can leave the cavities as a product of the catalytic reaction. Molecules with physical dimensions larger than the pore opening will be excluded from the cavities and will not be involved in the catalytic reactions. Catalysis products that are larger than the pore opening will be trapped in the cavities and undergo further catalysis. The cavities within the zeolite structures also affect the catalytic reaction because their size determines which transition-state compounds are formed. By choosing zeolites with the appropriate pore and cavity sizes, chemists can control the reactions that occur. Zeolites are particularly valuable for converting linear hydrocarbons into branched ones that have much higher octane ratings: linear hydrocarbons readily fit into the cavities, where they are rearranged into branched compounds. The cavities constrain the growth of these branched hydrocarbons to the desired size.
Maugh reported that catalysts may be made more selective by reducing the size of the catalyst from the conventional crystallites (microcrystals) to a well-defined cluster consisting of anywhere from two or three to a dozen or more metal atoms. Noble metals such as platinum, palladium, ruthenium, or rhodium deposited in the cavities of zeolites have been shown to improve the catalytic properties of zeolites.
Recently, Ozin reported a method by which zerovalent metal clusters could be formed within the intracrystalline cavities of some zeolites. The metal atoms are introduced as solvated metal atoms that can pass through the crystal structure and enter the alpha cages while maintaining the integrity of the zeolite structure. Metal clusters form when single atoms diffuse through the structure and agglomerate. The cluster sizes are controlled by the initial atom loading in the structure and the size of the alpha cage. This technique was proven to be a nondestructive means of depositing zerovalent metal atoms in the zeolite structure. Zeolites containing site-specific metal clusters may be potentially useful as catalysts for oil, gas, and petroleum production.
Economic Considerations
Natural zeolites have had only limited commercial success because of the widespread use of their synthetic counterparts before the existence of extensive deposits of natural zeolites was known. Preference was also given to synthetic zeolites because they are monomineralic; have a single type of cation with predictable ion-exchange capacities; have a given pore, channel, and cavity size with few stacking faults; and have few impurities.
The natural zeolites would seem to have great potential because of a single advantage, economy. There is an approximate 4-to-1 differential between the synthetic sodium A (Na A) available at $500/st and natural clinoptilolite at approximately $125/st. Comparison of natural versus synthetic zeolite prices is complicated by many factors. The Na A is produced by companies that do not have to recover research costs (the original patents have expired), and there is a very competitive market for the detergent builder business, considered the most promising large user. The price for the natural zeolites cannot be considered representative, either, as prices during a market development period can vary from high, reflecting the costs of custom mining, to low, reflecting a general market. Mining costs have not been established for zeolite mining operations in the United States.
Some of the inherent costs of synthetic zeolites cannot be quantified. Synthetic zeolites are produced in a slow, expensive, energy-consuming batch process. To achieve homogeneity of physical and chemical characteristics requires pure, raw materials, and there are considerable energy expenses in the synthesis and dehydration of synthetic zeolites, as well the tailoring processes for specific applications.
The natural zeolites, however, occur in layered sedimentary deposits mined from open pits. The overburden and the ore can usually by removed without blasting. Comminution of zeolites requires less energy than do other minerals, and the occurrence of zeolites in arid climates simplifies their dehydration. This provides an economic advantage over synthetic zeolites, which should permit their increased utilization in the mass markets where the costs of the synthetics would be prohibited.
An indepth analysis of the reasons for which the natural zeolite industry achieved lower success than expected was given by Eyde and Eyde in 1984. They indicated that zeolites are being improperly marketed. Natural zeolites are being sold as a replacement for materials that are in some cases both better for the use and cheaper than natural zeolites. To be successful, marketing approaches should treat the zeolites as “speciality chemicals” and address very specific markets that require their special properties. For example, zeolites have been used to remove radioactive cesium and strontium from low-level wastes from nuclear installations. They have also been used as dietary supplements for poultry and swine, as fertilizers to retain ammonium in the soil, and for stack-gas cleanup. These are uses that, while limited in volume, require continuous supplies of zeolites. Many uses are ideally suited for zeolites but represent little growth potential. One example is using clinoptilolite to remove ammonia from waste waters. Clinoptilolite was so efficient at removing ammonia from the effluent at waste water treatment plants that frequent recharging was unnecessary and the envisioned large and recurring market did not develop.
Applications
According to Mumpton, zeolites were first used 2,000 yr ago for building stones. The ion-exchange capability of zeolites was first investigated about 100 yr ago; the molecular sieving capability for separating gases, 40 yr ago; and the first commercial uses of synthetic zeolites, 30 yr ago. Breck reported that, in 25 yr, zeolites had achieved worldwide recognition as evidenced by the appearance of about 1,000 publications a year, and the recognition of 40 natural zeolite minerals and over 150 synthetic zeolites. Still, in 1977, only 10% of the known natural zeolites had commercial applications and fewer than 10% of the synthetic zeolites were successfully marketed.
Flanigen reported that the major use of natural zeolites is in bulk mineral applications: in Europe in the building and construction industry, where proximity to building location makes them cost effective, and in the Far East as filler in the paper industry, largely because of the unavailability of alternate mineral resources. A modest market for zeolite minerals has developed as a molecular sieve adsorbent in acid gas drying in the natural gas industry, in NH4 removal in water treatment systems by ion exchange, and in the production of oxygen and nitrogen via adsorptive air separation, especially in Japan. In general, however, their penetration into molecular sieve applications has been quite limited. Zeolite applications are summarized as follows:
Adsorption:
Regenerative processes:
Separations based on sieving.
Separations based on selectivity.
Purification.
Bulk separations.
Nonregenerative process:
Drying.
Windows.
Refrigerants.
Cryosorption.
Ion exchange:
Regenerative processes:
NH4 + removal.
Metal separations, removal from waste water.
Nonregenerative processes:
Radioisotope removal and storage.
Detergent builder.
Artificial kidney dialysate regeneration.
Aquaculture – NH4 + removal.
Ruminant feeding of nonprotein nitrogen (NPN).
Ion-exchange fertilizers. Catalysis:
Hydrocarbon conversion:
Alkylation.
Cracking.
Hydrocracking.
Isomerization,
Hydrogenation and dehydrogenation.
Hydrodealkylation.
Methanation.
Shape-selective reforming.
Dehydration.
Methanol to gasoline.
Organic catalysis.
Inorganic:
Hydrogen sulfide oxidation.
Ammonia reduction of nitric oxide.
Carbon monoxide oxidation.
H2O → O2 + H2.
Some experimental work was performed in the 1960’s using zeolites to remove cesium and strontium from water. Breck postulated that zeolites could be used commercially for the removal of the radioisotopes of cesium and strontium by ion exchange. In 1979, a mixture of synthetic and natural (chabazite) zeolites were used to remove those radioisotopes from contaminated water at the Three Mile Island nuclear power plant in Pennsylvania after cooling waters were contaminated with radioactive materials.
The specificity of clinoptilolite for ammonium has already been proven in water treatment, particularly for acquaculture, This specificity also may be useful for environmental control during uranium processing. Its purpose would be to adsorb ammonia that has contaminated ground water as the result of the leaching of uranium ores.
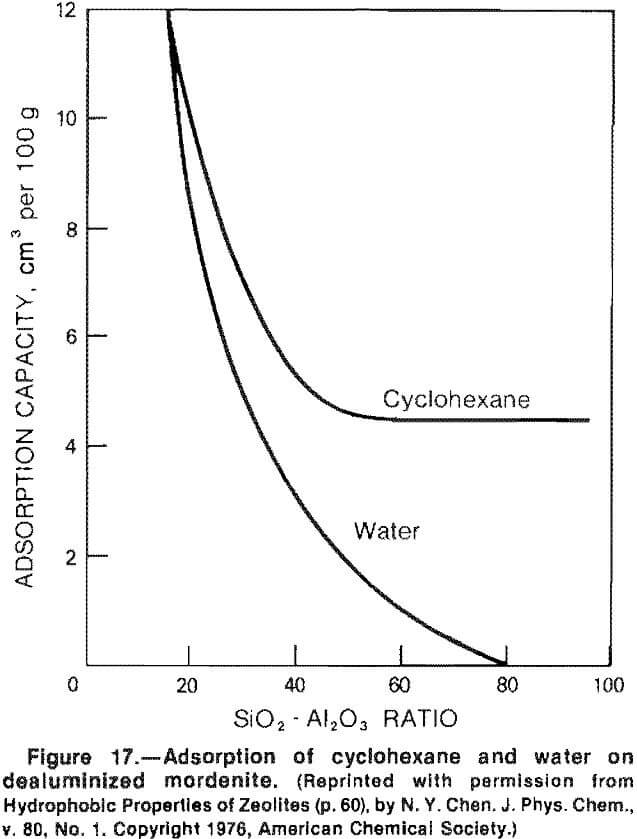
The natural zeolites have a pronounced selectivity for molecules with permanent dipoles, such as water. Reducing the silica-alumina ratio during synthesis or through acid leaching decreases the water absorption capacity, as shown in figure 17. Silicalite, the ZSM-5 molecular sieve polymorph, has no aluminum, has no active sites, and is hydrophobic. ZSM-5 can be formulated to contain enough aluminum to be active but little enough to remain hydrophobic. Such zeolites, then, can remove trace organics such as n-hexane from water but have greater stability during regeneration than the competing carbon.
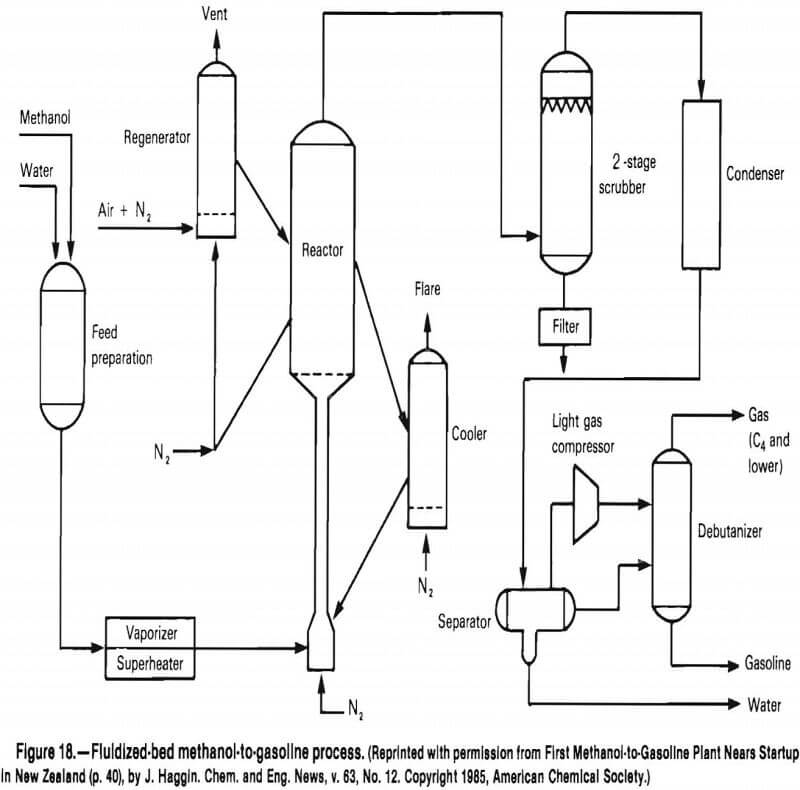
The Mobil Corp.’s methanol-to-gas (MTG) process using its ZSM-5 zeolite now has an operating plant in New Zealand. This plant has a fixed-bed reactor. The fluidized-bed process for MTG conversion has been declared successful after the completion of the final phase of operation at a pilot plant at Wesseling, Federal Republic of Germany. The raw gasoline yield is 90% with an octane rating of 90.25. The fluidized-bed process, shown in figure 18, is reported to significantly improve the MTG process and possibly permit feedstocks other than methanol to be used.
Meisel reported that Mobil Corp/s ZSM-5 catalyst is the most versatile of ail the shape-selective catalysts. He predicts many future application arising from its unique characteristics. He reports that it has all the right proper-
ties: catalytic activity, which may be tailored for a given reaction; exceptionally long life; and a shape-selective pore system whose channels closely match the size of molecules common to numerous petrochemical processes.
Applications for ZSM-5 catalyst range from the “building down” of molecules, the scrambling of molecules in various rearrangement processes, and the coupling of two different molecules, to the building up of smaller molecules to larger ones.
Meisel reported that 10 of the catalyst processes using ZSM-5 had been identified and developed in the short span of 10 yr. Five of these had been scaled up and were operating commercially in 35 reactors throughout the world. Several of the remaining five exhibited excellent commercial potential They collectively represent an unusual variety of refining and petrochemical processes and may reflect only a fraction of the potential of zeolite catalysts.
Besides the well-documented uses of zeolites as a desiccant, ammonium remover, and gas stream dryer, Garg and Ausikaitis reported the use of zeolites in energy-efficient adsorption cycle for removing large amounts of water (up to approximately 20%) from process streams. The adsorption cycle and the unique properties of zeolite molecular sieves can be used to dehydrate water-organic azeotropes. The dehydration of the ethanol-water azeotrope can be accomplished with less capital and lower energy costs [less than 2,000 Btu/gal (560 kJ/L)] using zeolites than with conventional azeotropic distillation methods. They claimed that this technology extends the use of molecular sieve adsorptive dehydration to the removal of over 20 wt % water from organic admixtures, and is capable, for example, of dehydrating the ethanol-water azeotrope from 190 proof (7.58 wt % water) to 199 proof ethanol.
In describing their process, Garg and Ausikaitis said that it uses a fraction of the energy required by conventional thermal-swing regeneration schemes. This energy savings is accomplished by operating in a manner that causes almost all of the heat of adsorption to be stored as a temperature rise within the bed during the adsorption step. The adsorption step uses the stored heat for regeneration energy and reduces the volume and temperature of the regeneration purge gas required.
Natural zeolities may prove a useful energy storage medium for solar energy for either cooling or heating. Energy, may be stored using a process that resembles a condensation-evaporation phenomenon. When gas is adsorbed on a solid, heat is always released, while the reverse process requires heat input. In a water-zeolite system, the zeolite is first dried by passing hot air from a solar collector through a container holding zeolite. The zeolite heats up, releasing adsorbed water, and the air becomes saturated with water. By using a suitable heat exchanger, this water vapor may be condensed and the heat of condensation extracted. After the zeolite has been dehydrated, it has the potential for producing heat. To extract heat, moist ambient air is driven through the zeolite bed. The water vapor in the air is adsorbed by the zeolite, releasing the heat of adsorption, with the result that warm dry air is produced. It is significant that even when the air is saturated with water vapor, airflow is never restricted. After the drying cycle, the zeolite may be allowed to cool down to room temperature to store energy indefinitely, provided that water vapor is excluded. This provides a decided advantage over other systems that require efficient insulation to maintain temperatures as high as possible even when heat is not being extracted,
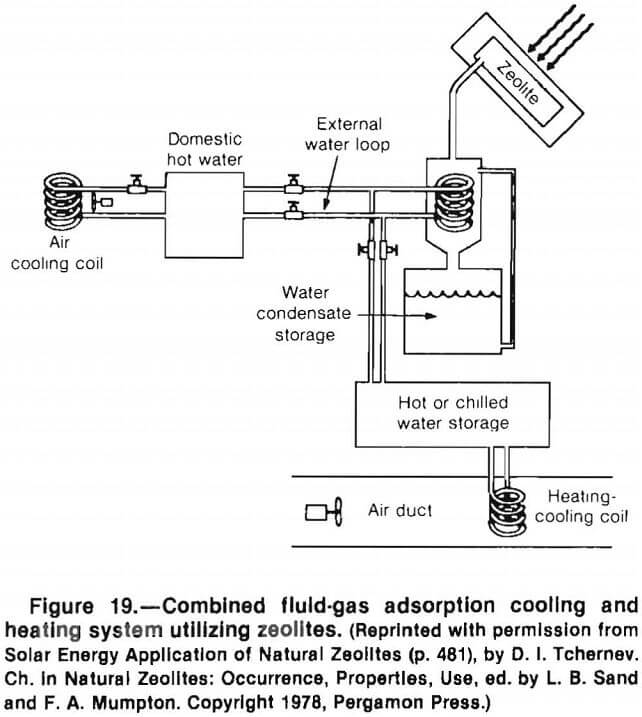
Tchernev explains that zeolites provide a unique opportunity to exploit solid-gas adsorption cooling systems because of their extremely nonlinear adsorption isotherms.
He had demonstrated the feasibility of using a zeolite system to provide domestic hot water and space heating with overall efficiencies above 75% and space cooling with an overall efficiency above 50%. The system uses natural chabazite or clinoptilolite as the solid adsorber and water vapor as the working fluid. Figure 19 is a diagram of his system.
In another zeolite-water interaction, a two-step thermo-chemical cycle for the decomposition of water using strong ionizing property of zeolites has been reported. Zeolites will induce electron transfer reactions between molecules and reduce multivalent cations of adsorbed molecules. A reaction cycle using the Cr³+-Cr²+ couple in mordenite was devised. Upon dehydration of the Cr³+ mordenite, reduction to Cr²+ occurs, with the evolution of oxygen. Upon rehydration, the divalent chromium oxidizes to Cr³+ with the evolution of hydrogen. Similar results were obtained with an In³+ exchanged mordenite. The zeolite used was a hydrogen form of synthetic mordenite.
The initial major uses for natural zeolites may be for agriculture-aquaculture areas. The economic advantages of the natural minerals for large tonnage uses would seem to dictate their usage here. The present status in that area was best expressed by Mumpton in the abstract of his keynote address at the international conference, “Zeo- Agriculture ’82”:
Following earlier work in Japan and other countries numerous studies have been carried out in recent years in the United States and abroad on the agriculture potential of zeolite minerals with considerable success, and with sortie failures. Based on their attractive ion-exchange, adsorption, and hydration properties, natural zeolites have been found to act as slow-release fertilizers to provide potassium and nitrogen to agricultural soils, as carriers of herbicides, fungicides, and insecticides, as possible traps for heavy metal contaminants in soils amended with municipal sewage, and as decaking agents for fertilizer and feed storage. Their addition to the normal diets of swine, poultry, and ruminants has resulted in increased body weights and feed efficiencies, with simultaneous decrease in the incidence of scours and other intestinal diseases, especially among young animals.
Zeolites have also been shown to deodorize and increase the nutrient content of animal excrement, thereby improving the fertilizer value of the manure and lessening the energy required for ventilation of poultry houses and other buildings for confined livestock Zeolite filtration has been employed to remove toxic concentrations of ammonia from aquacultural systems, and zeolite adsorption processes have been, developed to provide oxygen-enriched air for such systems, both of which allow more fish to be raised or transported in the same volume of water. Preliminary experiments even suggest that adding zeolites to fish rations can increase biomass production. The hydration-dehydration properties of certain zeolites have been harnessed in the development of solar-refrigeration units to preserve dairy and other animal products and veterinary supplies in areas where electricity is not readily available.
Although many of these investigations are preliminary, and many have failed to reproduce the results obtained in other laboratories, the experimental evidence to dale strongly suggests that continued investigation under controlled conditions will lead to numerous areas where natural zeolites can contribute to increased animal and crop production. The adage that “where there’s smoke, there’s fire” will undoubtedly hold true for the emerging field of zeo-agriculture.
Flanigen reported that “future trends in materials will no doubt see the development of new commercial zeolites selected from newly discovered compositions and structures, chemical modifications of present commercial products to generate new and useful properties, and a reevaluation of the host of known zeolites which never achieved commercial success. It seems likely that, with the increasing number of laboratories devoting resources to the search for new structures and compositions, new classes of molecular sieve materials will be discovered. The modification chemistry of zeolites, practiced to date, such as steaming and chemical extraction, leaves a vast area of chemical and structural modification of solids as yet unexplored with zeolites.”
She also predicts that “additional types of natural zeolites will probably not achieve commercial prominence since the large geological exploration efforts for zeolite deposits throughout the world during the last ten to fifteen years have probably identified all of the zeolite mineral species of commercial significance” and that “natural zeolites should continue to grow as an important industrial mineral resource used principally in bulk application areas. The ‘engineering” of the mined zeolites, by beneficiation to upgrade purity and chemical modification to tailor properties, will no doubt emerge as the level of technically intensive effort on natural zeolites expands.”
As a last note, it is interesting that the creators of the synthetic zeolite industry have themselves created a competitive product for at least the molecular sieve portion of their market. The Union Carbide Corp. scientists learned enough about porous crystals from their pioneering zeolite research to enable them to discover and develop a family of aluminophosphate and silica-aluminum phosphate crystalline solids. These microporous solids have slightly different physical and chemical properties from zeolites and have potential uses as molecular sieves.
Summary
Zeolites are crystalline aluminosilicates of the alkaline and alkaline-earth metals. They possess many desirable ion- exchange, molecular sieving, and catalytic properties, which make them valuable mineral commodities. It is only within the past 40 yr that zeolites have been used extensively in commercial processes. The petroleum industry, for example, has profited immensely from the use of zeolites in refining operations. Ironically, research on the properties of natural zeolites created the synthetic zeolite industry. Commercial applications for natural zeolites, which were considered to be mineralogical curiosities, were discovered, so the emphasis during the 1940’s and 1950’s was on the commercial production of their synthetic counterparts. Synthetic zeolites were firmly established in the market when large deposits of natural zeolites were discovered in the 1950’s and 1960’s. Only recently have natural zeolites begun to be used commercially, not as replacements for synthetic-zeolites but in areas where the use of synthetic zeolites would prove uneconomical.