
Table of Contents
Bacterial sulfate reduction has been identified as one process by which constructed wetland systems remove contaminant metals from coal and metal- mine drainage. Under anaerobic conditions, sulfate-reducing bacteria oxidize simple organic compounds (CH2O) with sulfate, and thereby generate hydrogen sulfide and bicarbonate ions:
SO4-² + 2CH2O → H2S + 2HCO3-…………………………………………..(1)
Hydrogen sulfide reacts with many contaminant metals to form insoluble metal sulfides:
H2S + M+² → MS + 2H+…………………………………………………….(2)
where M includes metals such as Fe, Zn, Mn, Ni, Cd, Cu, and Pb. Bicarbonate ions can consume protons to raise the pH of acidic water:
HCO3- + H+ → CO2 + H2O…………………………………………………..(3)
Most wetlands constructed with an organic substrate and receiving sulfate- enriched mine drainage develop an anaerobic zone in which sulfate reduction becomes established. However, the main flow path of water in these wetlands bypasses the anaerobic zone and is therefore minimally affected by sulfate reduction. As an alternative, the entire flow of contaminated water could be directed through enclosed reactors, within which an organic-rich, anaerobic environment favorable for sulfate reduction is maintained. The design of such a reactor system for treating metal- contaminated water requires:
- exclusion of oxygen,
- a source of sulfate (generally the contaminated water),
- a source of simple organic compounds to serve as a bacterial carbon source, and
- some way to physically retain the metal sulfide precipitates within the system.
Since low pH (<5) inhibits sulfate-reducing bacteria (Postgate 1984), and increases the solubility of metal sulfides, a sulfate reduction system must also be designed to generate sufficient alkalinity to buffer the pH of acidic inflows.
Hammack and Hedin (1989) and McIntire et al (1990) studied simple sulfate reduction reactors consisting of carboys or columns filled with organic matter. They found that their reactors lowered iron, nickel, and manganese concentrations in synthetic mine drainage by 60 to 90%. These results led us to construct pilot-scale reactor systems of a similar design, and to study their potential for treating metal-contaminated water. In this paper we present our initial findings relating to the performance capabilities and chemistry of the pilot-scale reactors, and discuss strategies for optimizing their metal-retaining and alkalinity-generating potentials.
Methods
Two pilot-scale reactor systems were constructed in 1990. The reactors in each system consisted of either capped barrels (Pittsburgh system) or covered tanks (Palmerton system), that were filled with loosely-packed spent mushroom compost. Spent mushroom compost consists of a composted mixture of manure, hay, straw, corn cobs, and wood chips that has been conditioned with gypsum and limestone, and used for cultivating mushrooms. Since it decomposes readily, the compost served as a source of organic carbon for bacterial sulfate reduction, and its bulk form served to physically detain any precipitated solids. Selected properties of the different composts used in each reactor system are shown in Table 1.
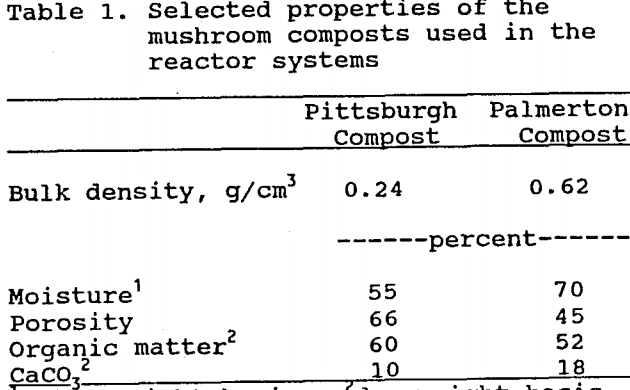
The Pittsburgh reactor system was installed to treat acidic, iron-contaminated drainage within the Experimental Mine at the U.S. Bureau of Mines, Pittsburgh Research Center. This system consisted of three 200 L reactor barrels plumbed in series and receiving water from a 1140 L supply tank (Fig. 1). Barrels were used instead of a single large reactor because of height limitations within the coal mine. The underground location of this system maintained a nearly constant ambient temperature of 10°C, which permitted operation during the winter.
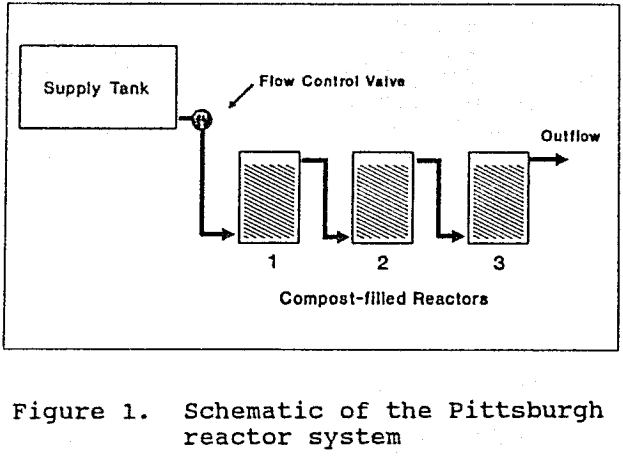
The Palmerton reactor system was installed to treat metal-contaminated drainage from a smelting residues dump at the former New Jersey Zinc Company plant in Palmerton, Pennsylvania. This system consisted of two independent, 4500 L reactor tanks receiving water from a 3500 L supply tank (Fig. 2). The data presented for this system were collected during summer operation, when average ambient temperature ranged between 18 and
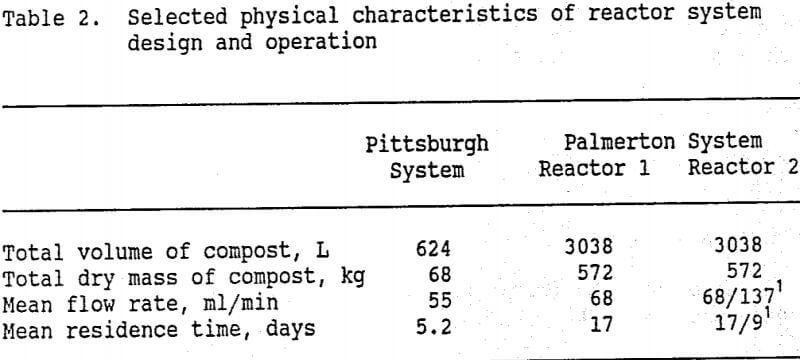
Flow through the reactor systems was by gravity and was controlled by valves in the inflow plumbing. Flow rates were measured and adjusted twice per week, and residence times were calculated from these rates. Our primary objective was to evaluate the chemistry and water treatment potential of the reactor systems. Therefore, we used residence times at which we expected significant metals removal to occur. In the Palmerton system, the flow rate through one of the reactors (Reactor 2) was doubled after ten weeks of operation to observe the effects of a shortened residence time. Table 2 shows the quantity of compost contained in each system, and the mean flow rates and residence times at which each system was operated.
Water Sampling and Analysis
At 1 – 2 week intervals, influent samples were collected from the supply tanks and effluent samples were collected from the outflows of each system. Samples
of pore water were occasionally collected from the Pittsburgh system by inserting a 5-mm diameter tube into the compost to the desired depth and using a syringe to draw a sample. Oxidation and hydrolysis reactions within the Pittsburgh system supply tank caused influent iron and aluminum concentrations to vary over short intervals (<1 week). To assure that influent and effluent samples could be compared directly, we therefore set the collection interval equal to system residence time. This problem with influent stability was not encountered in the Palmerton system, nor with ions other than iron and aluminum in the Pittsburgh system.
Raw samples were collected for determination of pH, alkalinity, and anions other than sulfide. Samples for dissolved sulfide analysis were treated upon collection with 5 ml of 1N NaOH/250 ml of sample to prevent outgassing of H2S. Samples for total metals analysis were treated upon collection with 3 ml of acid/250 ml of sample to keep the metal ions in solution. Concentrated hydrochloric acid was used for the Pittsburgh system, to avoid oxidizing ferrous iron, and 70% nitric acid was used for the Palmerton system. samples for dissolved metals analysis were filtered through a 1.2 µm glass fiber filter prior to preservation with acid.
Water samples from both systems were analyzed at the U.S. Bureau of Mines, Pittsburgh Research Center (PRC), where samples were received from the Pittsburgh system within one hour of collection, and from the Palmerton system after one week of unrefrigerated shipment. In addition, duplicates of the Palmerton system samples were analyzed at the Horsehead Resource Development Company (HRD) laboratory in Palmerton, Pennsylvania, where the samples were received on the day of collection. At both laboratories, the samples were stored refrigerated, and analyzed within one to two weeks of receipt. Dissolved sulfide was measured only at the PRC laboratory, and usually on the same day the samples were received. Sample pH was determined at collection time for the Palmerton system, and ordinarily at analysis time for the Pittsburgh system.
Prior to metals analysis, samples from the Pittsburgh system were filtered or decanted to remove precipitated humic acids, whereas samples from the Palmerton system were boiled with nitric acid until colorless. Atomic absorption spectroscopy was used by the PRC laboratory to determine K and Cd concentrations, and by the HRD laboratory to determine Na and Zn concentrations. Ferrous iron was determined by titration with K2Cr2O7. In all other cases, metals were determined using an ICP atomic emission spectrophotometer. Sulfate was measured by the PRC laboratory using a liquid chromatograph coupled with a conductivity detector, and by the HRD laboratory using the gravimetric BaCl2 method. Alkalinity was determined by titrating a sample with 0.2N H2SO4 to pH 4.5. Sulfide was measured using an ion-specific electrode.
The results from the PRC and HRD laboratories were generally within 20% of one another, so the data from the two laboratories were averaged for the Palmerton system.
Compost Sampling and Analysis
The spent mushroom compost used in the Pittsburgh system was sampled before being placed in the reactor barrels, and after 18 weeks of system operation. Compost from the Palmerton system was similarly sampled, but the analyses were not completed in time for this paper. The 18-week samples from the Pittsburgh system were collected from depths of 10 cm and 60 cm in each of the three reactor barrels (maximum depth = 75 cm). At each depth, a single composite sample of three to five subsamples was collected by hand, and immediately sealed in an argon-filled container. All samples were refrigerated within one hour of collection.
The total content of inorganic cations and anions was determined by ashing subsamples in a muffle furnace at 550°C for six hours, boiling the ash in concentrated HCl, and analyzing the acid extract by the methods previously described for water samples. A residual fraction consisting primarily of silica and clay minerals remained undissolved by this procedure. The moisture content of each sample was estimated by drying separate subsamples to constant weight at 112°C.
The total iron content was expected to consist of a “fixed” fraction, tightly bound within organic matter and resistant clay minerals, and a “labile” fraction, occurring in forms, such as FeOOH, that are soluble under reducing or acidic conditions. Because of its potential solubility, the labile iron would be susceptible to leaching, and would be available for reaction with H2S. The labile iron content of the compost samples was determined by anaerobically extracting subsamples with 4N HCl under an argon sparge, and analyzing the extract for iron.
A single subsample from the 10 cm depth in the first reactor barrel was examined by Mossbauer spectroscopy (Willard et al 1974) to directly identify the chemical forms of iron present.
Acid-volatile sulfide (AVS), consisting of HS-, H2S, and FeS, was determined using an anaerobic distillation procedure (Herlihy and Mills 1985), in which the sulfide was liberated as H2S with concentrated HCl, stripped from the subsamples with argon, and collected in NaOH traps for sulfide analysis. Chromium-reducible sulfur (CRS), consisting of pyrite (FeS2) and elemental sulfur, was determined using a similar procedure in which subsamples, already extracted for AVS, were boiled with HCl to which Cr+² ion was added to reduce these remaining sulfur forms to H2S. The AVS and CRS analyses were run within two hours after the compost samples were collected.
Calcium carbonate content was deduced from the results of the ashing/acid extraction analysis described above. The only two species present in the ash but not measured in the extract were believed to be oxide and carbonate. Therefore, the portion of ash weight not accounted for by ions measured in the extract was apportioned between oxide and carbonate, such that an exact charge balance was attained between measured cations, and measured anions plus estimated oxide and carbonate. The estimated carbonate was assumed to have been present in the compost before ashing, and to have been associated solely with calcium, as CaCO3.
Results
Following start-up, both systems underwent an initial phase during which Ca, Mg, Na, K, and sulfate ions were leached from the compost. By the time this phase was completed, effluent color had changed from dark brown to yellow, the effluent acquired a strong hydrogen sulfide odor, effluent base-cation concentrations (except calcium) had fallen to influent levels, and net sulfate consumption (reduction) and metal retention had become discernible. The initial leaching phase was complete for the Pittsburgh system after 3.5 weeks of operation and 3940 L of flow, and for the Palmerton system after 6 weeks of operation and 4140 L of flow.
The results presented below pertain to system performance following the initial leaching phase. For the Pittsburgh system, a 14-week period involving 6110 L of flow is considered, and for the Palmerton system, a 6-week period involving 4210 L (Reactor 1) to 5680 L (Reactor 2) of flow is considered.
Changes in Water chemistry
Mean influent and effluent water analysis results are shown for the Pittsburgh system in Table 3, and for the Palmerton system in Table 4. Significant concentrations of Fe and Al occurred only in water samples from the Pittsburgh system, whereas Zn, Mn, Ni, and Cd were important only in the Palmerton system samples. Both systems lowered contaminant metal concentrations by greater than 95%, lowered sulfate concentrations by about 30%, and produced circumneutral effluents having high alkalinity. The effluents from both systems contained high concentrations of dissolved sulfide (HS- and H2S), which, together with the lowering of sulfate concentrations, indicates that bacterial sulfate reduction was occurring. Both systems continued to release calcium after leaching of the other base cations had ceased. Possible origins for this calcium include the dissolution of limestone (a constituent of the spent mushroom compost) and the decomposition of calcium-rich organic matter.
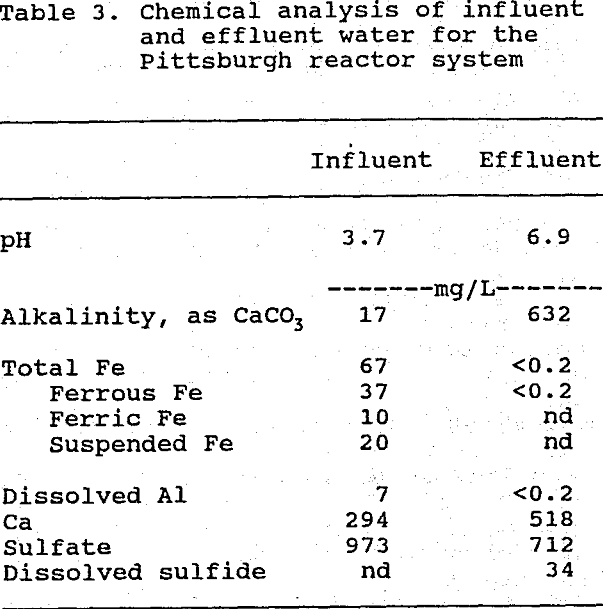
Values are means of 8 influent and 11 effluent samples; nd = not detectible;
Alkalinity was only present in samples with pH >4.5 (n = 2).
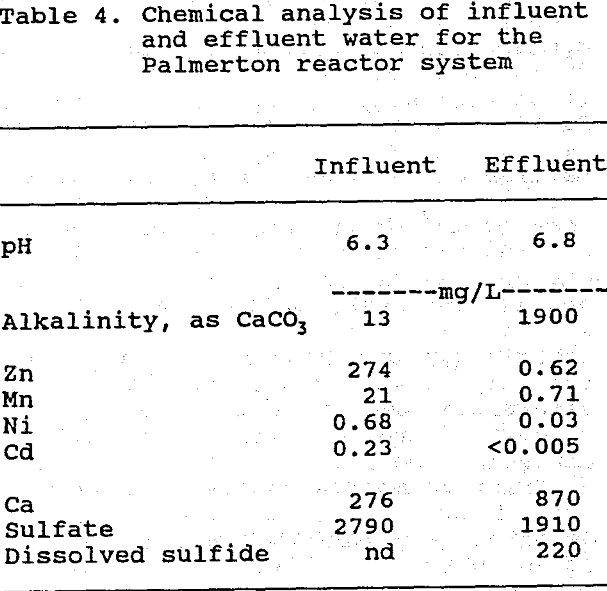
Values are means of 6 influent and 12 effluent samples; nd = not detectable
The Pittsburgh system consistently lowered aluminum concentrations to less than 0.2 mg/L. Since aluminum does not form a stable sulfide in the presence of water, this removal likely resulted from its hydrolysis to insoluble Al(OH)3:
Al+³ + 3H2O → Al(OH)3 + 3H+…………………………………………………(4)
This reaction is spontaneous at the circumneutral conditions that prevailed within the reactor barrels (Stumm and Morgan 1981). Ferric iron also hydrolyzes at circumneutral pH, but in the anaerobic environment within the barrels, it apparently was reduced to ferrous iron, which requires a much higher pH for hydrolysis. This reduction probably occurred in conjunction with the microbial oxidation of organic matter:
4Fe+³ + CH2O + H2O → 4Fe + CO2 + 4H+………………………………………(5)
Ferric iron was never detected in water that had passed beyond 7% of the total system volume, though ferrous iron concentrations usually remained high beyond that point. Also, suspended ferric oxyhydroxide (FeOOH) did not visibly accumulate in the inflow zone of the first reactor barrel, which suggests that influent FeOOH was similarly reduced and dissolved:
4 FeOOH + CH2O + 7CO2 + H2O → 4Fe+² + 8HCO3-……………………………..(6)
Analyses of pore water from the Pittsburgh system revealed that essentially all of the influent iron was removed by the first reactor barrel. Also, very little dissolved sulfide and alkalinity left the first barrel, thereby leaving the second and third reactor barrels to generate the high levels of dissolved sulfide and alkalinity observed in the system effluent.
Changes in Compost Composition
Table 5 shows the results of the analyses of Pittsburgh system compost. The total iron content for the first and second reactor barrels did not change significantly during the course of this study. This is consistent with the results of the water analyses, which detected very little iron leaching from the system, and which indicated that the influent iron was being removed entirely in the first barrel. Since little iron left the system, most of the original 63 µmoles of labile Fe/g compost was retained within the reactors.
Comparison of the total iron contents indicates that the first reactor barrel accumulated 90 µmoles of Fe/g compost from the influent water. By adding this accumulated iron to the 63 µmoles/g of retained labile iron, a total iron retention value of 153 µmoles/g is obtained. This closely matches the acid-volatile sulfide (AVS) content of 150 µmoles/g that was found in the first barrel, thus suggesting that iron was retained primarily as ferrous monosulfide (FeS). In agreement with this, Mossbauer spectroscopy analysis identified FeS as the predominant iron form present.
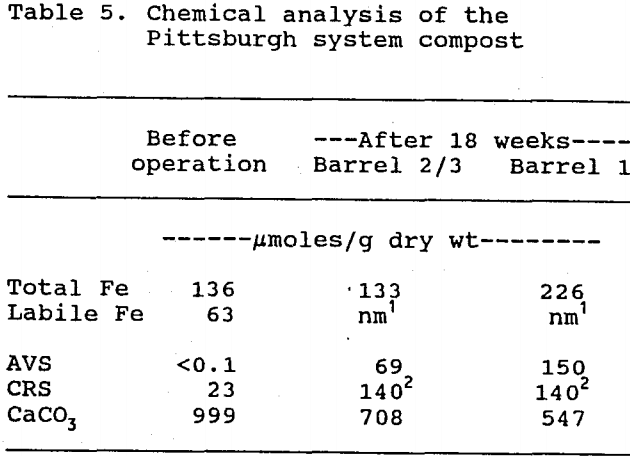
Note that, despite the absence of net iron accumulation in the second and third reactor barrels, AVS nevertheless accumulated there, evidently due to the reaction of H2S with the labile iron in those barrels.
Of the total accumulation of reduced sulfur (AVS and CRS) within the system, an average of 55% was accumulated in the chromium-reducible sulfur (CRS) fraction. Mossbauer spectroscopy failed to detect any pyrite (FeS2) in the compost, which suggests that elemental sulfur was the CRS form accumulated.
During system operation, the limestone content in the compost decreased by 35% for the entire system, and by 45% in the first barrel. This decrease confirmed the hypothesis that limestone dissolution generated much of the system’s net effluent calcium and alkalinity:
CaCO3 + CO2 + H2O → Ca+² + 2HCO3…………………………………………….(7)
Limestone dissolution was evident in the second and third reactor barrels, where pore water pH was circumneutral, as well as in the first barrel, where pore water pH was often lower. The mean rate of limestone dissolution for the system was 234 mg CaCO3/L of flow.
Sulfur Budget
The results of the water and compost analyses identified three sinks for the hydrogen sulfide produced by bacterial sulfate reduction: reaction with metal ions to form insoluble metal sulfides (AVS), conversion to CRS forms, and escape from the system in the effluent as unreacted sulfide. Figure 3 shows the mmol/L of sulfate reduced (total bar height) over time for the Pittsburgh
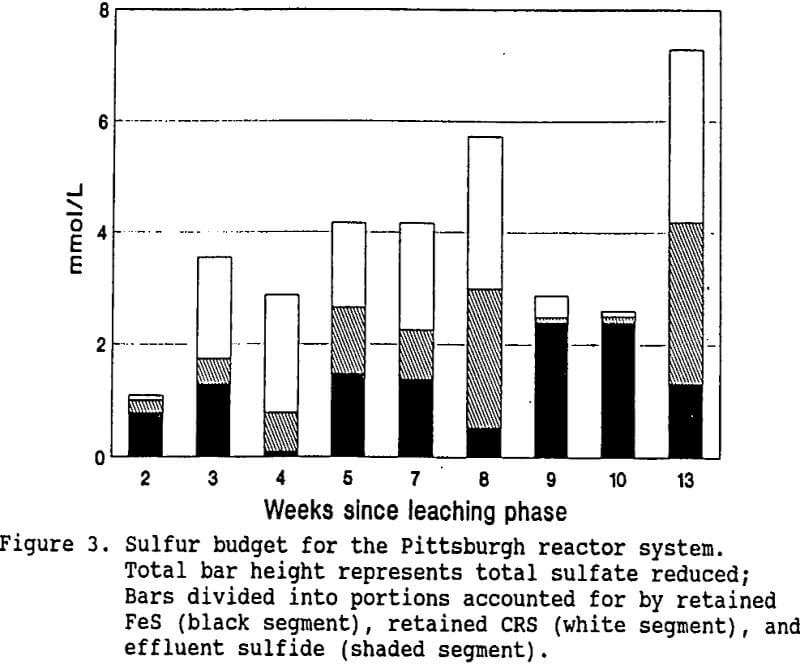
system, divided according to the portions accounted for by iron accumulation, effluent sulfide, and CRS (by difference). All of the sulfate removed from the influent water was assumed to have been reduced to H2S. Since the influent iron was completely retained by the system, the bar segment representing iron retention also represents influent iron concentration. Note that for weeks 4 and 8, when influent iron concentrations were less than 0.5 mmol/L (28 mg/L), over four times as much sulfide escaped in the effluent as was retained as FeS. In contrast, during weeks 9 and 10, when influent iron concentrations were around 2.4 mmol/L (134 mg/L), nearly all of the sulfur reduced was retained as FeS, and very little effluent sulfide escaped.
Table 6 shows the mean rates of sulfate reduction, metal retention, and AVS/CRS accumulation computed for the two reactor systems. The iron retention and AVS/CRS accumulation rates for the Pittsburgh system were computed using compost analysis data:
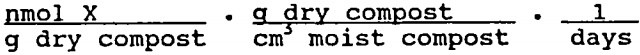
The other rates were computed from water analysis data:
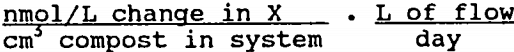
The rates of AVS accumulation almost equal those of total Fe retention for the
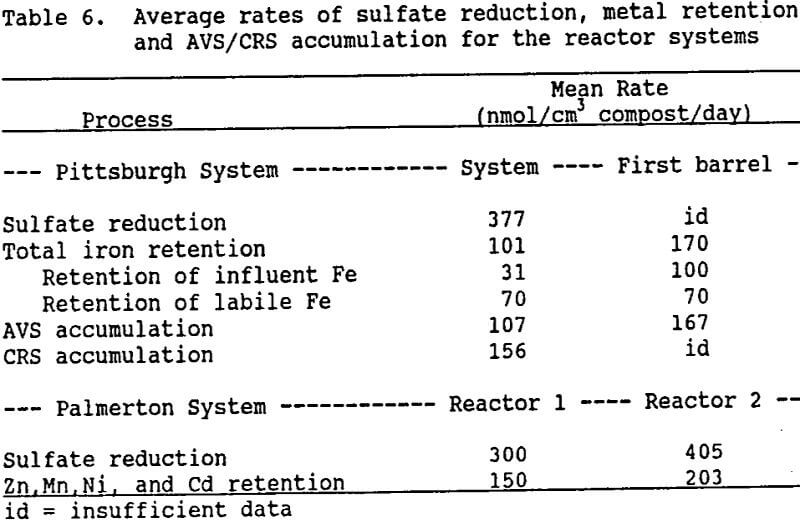
Pittsburgh system, as would be expected if FeS was being formed. Note that the sulfate reduction rate for the Pittsburgh system exceeds the sum of the AVS and CRS accumulation rates by 114 nmol/cm³/day. This difference suggests an average effluent sulfide concentration of 37 mg/L, which is close to the measured average of 34 mg/L. By neglecting the possibility of CRS formation, and similarly computing effluent sulfide from the Palmerton system rates, a value of 14 6 mg/L sulfide is obtained for both Reactor 1 and Reactor 2. This concentration is low in comparison with the measured average of 220 mg/L, which indicates that more sulfide escaped the Palmerton system unreacted than the metal retention rates suggest. Thus, some of the metal removal observed in the Palmerton system apparently occurred through processes other than sulfide precipitation.
Origins of Effluent Alkalinity
Both reactor systems produced effluents having high alkalinity. For the Pittsburgh system, chemical equilibrium calculations indicate that this alkalinity consisted of 1.1% HPO4-², 4.5% HS-, and 94.4% HCO3- plus organic anions. Corresponding values calculated for the Palmerton system are 1.3%, 12.5%, and 84.2%, respectively. The reactions
identified from the water and compost analyses as being important in the reactors (Equations 1, 2, and 4 – 7) produce alkalinity (as HCO3-) or acidity (as H+). These equations were used to derive the following equation for predicting effluent alkalinity (meq/L) from measured changes in the concentrations (mmol/L) of SO4-², Ca+², FeOOH, M+², Al+³, and Fe+³
Alkalinity = 2SO4-² + 2Ca+² + 2FeOOH
– 2M+² – 3Al+³ – 1Fe+³
+ influent alkalinity
– influent H+ (8).
The derivation of Equation 8 assumes that Equations 1, 2, and 4-7 are the sole sources of alkalinity and acidity, and that the concentration changes observed for the indicator ions are induced solely by the reactions they indicate.
Figure 4 shows the alkalinity values computed using Equation 8, along with the corresponding measured alkalinity values for the reactor system effluents. The agreement between the two sets of values supports the hypothesized origins of effluent alkalinity. Correlation of the two sets of values gives an r² of 0.92 (P < .01; n = 8) for the Pittsburgh system, and 0.56 (not significant; n = 6) for the Palmerton system. The weaker correlation of the Palmerton values may be due to the previously mentioned removal of metals in non-sulfide forms. This non-sulfide removal would mean that Equation 2 proceeded to a lesser extent than the change in metal concentration (M) indicates.
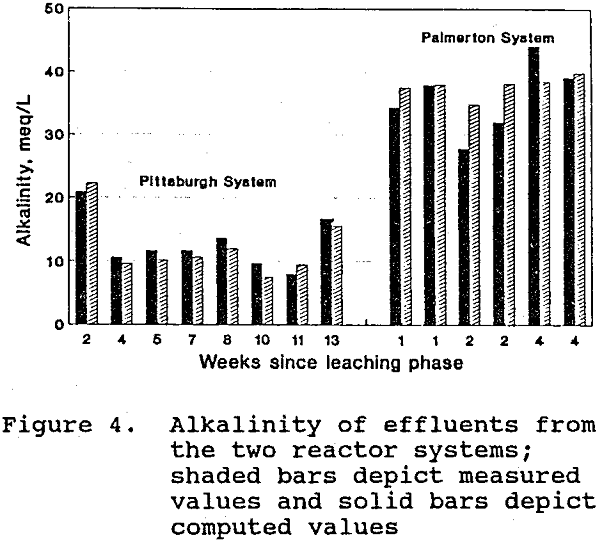
Sulfate reduction (Equation 1) and limestone dissolution (Equation 7) were the dominant sources of alkalinity for both systems. Of the total computed alkalinity generated by these two reactions combined, limestone dissolution contributed an average of 53% for the Pittsburgh system and 61% for the Palmerton system.
Discussion
The results presented in this paper confirm that the reactors precipitated contaminant metals as insoluble sulfides following their reaction with biologically-generated hydrogen sulfide. Several strategies for enhancing the metal-retaining and alkalinity-generating effects are suggested by the results.
The maximum quantity of metal that can be precipitated in a sulfide form is dictated by the quantity of H2S available for reaction. One means of enhancing metal retention therefore might be to increase the rate of bacterial sulfate reduction. McIntire et al (1990) found that when lactate, a preferred carbon source for sulfate-reducing bacteria, was added to the metal-contaminated water entering their reactors, sulfate reduction and metal retention increased. This suggests that the type or quantity of organic compounds released by the decomposing spent mushroom compost limit the rate of sulfate reduction. Thus, the rate might be increased by filling the reactors with a different organic material, or by supplementing the compost by adding organic compounds to the influent water. Since sulfate reduction is a microbial process, its rate might also be increased by using higher flow rates to prevent the possible accumulation of inhibitory compounds, or by raising reactor temperature.
Another means of enhancing reactor metal retention would be to modify reactor design or operation so that all of the H2S produced reacts completely to form metal sulfides and none is lost as effluent sulfide or CRS. For the Pittsburgh system, an inverse relationship was found between influent iron and effluent sulfide concentrations. Similarly, pore water sulfide concentrations were low where dissolved iron was still present, and high where dissolved iron was absent. These observations demonstrate that excess sulfide, destined for release in the effluent, can be exploited for metals removal, provided the system is fed metal ions at a sufficiently high rate. For metals that are relatively toxic to bacteria, such as cadmium and copper (McIntire et al 1990; Postgate 1984), the influent metal load might best be adjusted by modifying flow rate, whereas, for other metals, influent concentration could also be modified. No means can be suggested for exploiting the H2S converted to CRS forms, since the pathway of this conversion is poorly understood.
If acidic water is to be treated, the reactors must generate net alkalinity to prevent the bacteria from being inhibited and metal sulfides from being dissolved. Limestone dissolution in the reactors generated slightly more alkalinity than did sulfate reduction. When the compost limestone content eventually becomes depleted, or in a reactor built without limestone, sulfate reduction would have to be relied upon as the principal alkalinity generator. Since metal sulfide formation (Equation 2) consumes as many moles of alkalinity as are generated by sulfate reduction (Equation 1), no net alkalinity will be gained from sulfate reduction if all of the H2S produced is consumed for metal retention. Thus, sulfate reduction serves as an important alkalinity source only if some H2S remains unreacted. In the Pittsburgh system, this tradeoff between alkalinity generation and H2S consumption was observed directly, as most of the net alkalinity and excess H2S accumulated in the second and third reactor barrels in the absence of iron removal.
This study demonstrates that sulfate reduction reactors of the design studied here are capable of removing Fe, Zn, Mn, Ni, and Cd from contaminated water. Without modifying reactor design, metal retention rates can be maximized by providing influent metal loads sufficiently large to completely consume the H2S generated. The theoretical maximum metal retention rate will equal the sulfate reduction rate. If acidic water is being treated, the reactor interior must be protected from acidification, either by incorporating limestone into the organic substrate, or by operating the reactors at a lower metal retention rate such that H2S is produced in excess of the amount that will be consumed by metal sulfide formation. Higher rates of metal retention could also be attained by modifying reactor design to encourage higher rates of bacterial sulfate reduction.