
Table of Contents
Present copper extraction technology is based principally on pyrometallurgical processing of sulfide concentrates in which high-temperature air-roasting and air-smelting generates large quantities of sulfur dioxide. Because of very stringent environmental standards and increasing cost of energy, there has been a renewed interest in hydrometallurgical processes for recovering copper from sulfide flotation concentrates. Potential advantages of this alternate approach are (1) lower capital investments because smaller plants are feasible, (2) increased flexibility in handling various grades of concentrates, (3) recovery of elemental sulfur and iron oxide, and (4) possible reduction in energy requirements.
This report is limited to a discussion of a novel approach that combines a preliminary inert gas roast with a two-stage leach plus electrowinning. The principal steps consist of inert gas roasting to convert the chalcopyrite to sulfur-deficient chalcopyrite, troilite, and a bornitelike structure; hydrochloric acid leaching to remove most of the iron; then dissolving the copper-bearing residue in an oxidizing spent electrolyte followed by electrowinning. The products are high-purity copper, elemental sulfur, iron oxide, and a silica residue containing gold, silver, and molybdenum.
Fundamentals
The objective of the roasting step is to transform chalcopyrite into crystallographic phases suitable for selective leaching of the iron. During the inert gas roasting step, approximately 20 percent of the sulfur is removed and is collected in the flue dust as elemental sulfur together with some of the more volatile impurities such as arsenic, bismuth, lead, and zinc.
The next operation is to leach the calcine with hydrochloric acid to remove most of the iron leaving a residue of digenite (Cu8 S5), undissolved modified chalcopyrite, and hydrochloric acid-insoluble minerals such as quartz and molybdenite together with precious metals. Removal of iron at this stage is necessary to avoid a prohibitively high buildup of iron during the copper electrowinning step. Hydrogen sulfide liberated during hydrochloric acid leaching of the calcine converts any dissolved copper to insoluble copper sulfide. Any excess hydrogen sulfide can either be burned to form sulfur dioxide for use in the electrowinning step or reacted with sulfur dioxide to form elemental sulfur. This sulfur dioxide could be obtained by oxidizing part of the hydrogen sulfide. The hydrogen sulfide-sulfur dioxide mixture then would react to give elemental sulfur.
The digenite in the residue from the hydrochloric acid leach is dissolved in a spent electrolyte recycled from the copper electrowinning step. The presence of ferric ion in the CuSO4·H2SO4·FeSO4 electrolyte is essential for the dissolution of digenite. Oxygen is fed into the second-stage leach solution to continuously reoxidize the ferrous iron back to the ferric state.
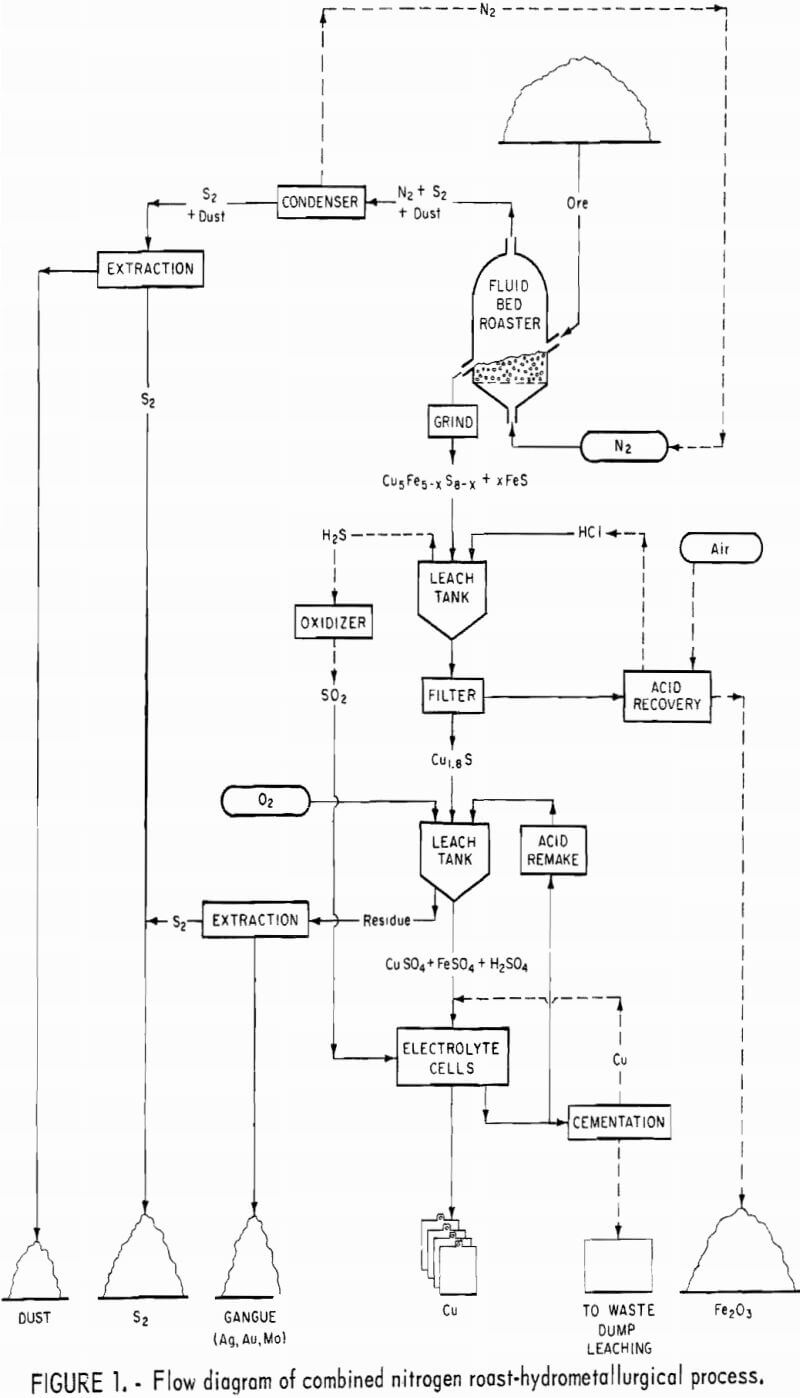
The leach solution, now enriched in copper -30 grams per liter, then is subjected to electrowinning to recover the copper and to regenerate spent electrolyte for dissolving more digenite. The silica residue remaining from the second-stage leach contains the insoluble precious metals, molybdenum as molybdenite, and elemental sulfur formed during the dissolution of digenite. A flow diagram for the combined pyrometallurgical-hydrometallurgical process is shown in figure 1. Steps on the flow diagram that were not investigated in this study are shown with dotted lines.
Samples
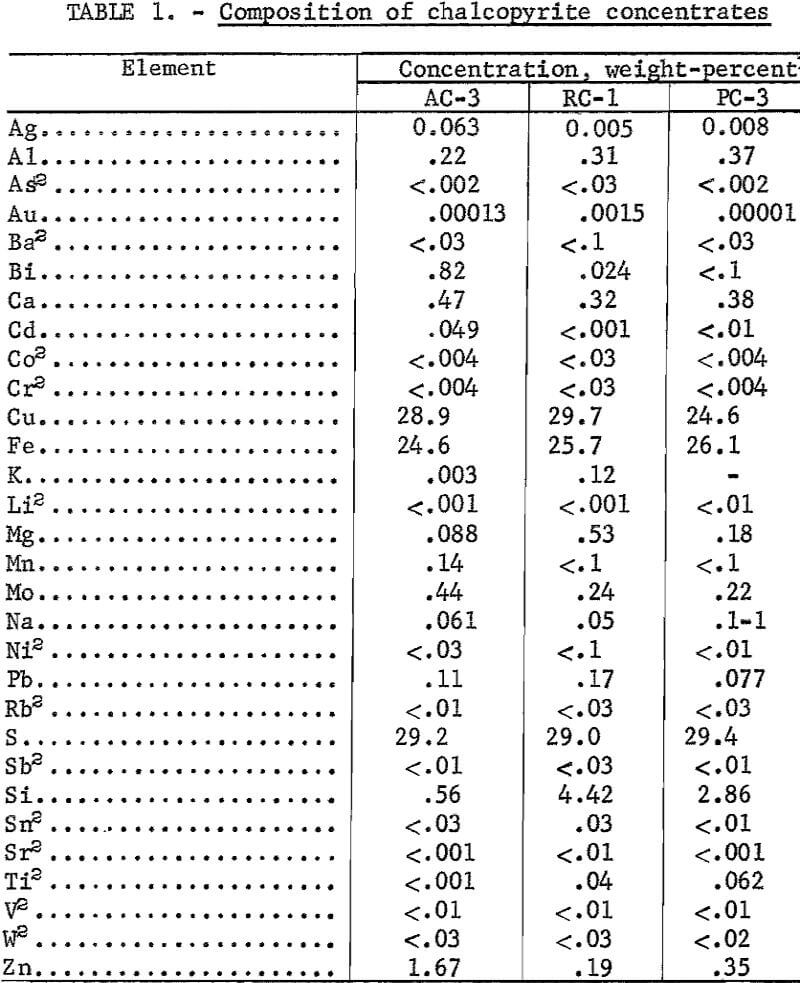
Twenty-five- to fifty-pound quantities of chalcopyrite concentrates were obtained from three commercial smelters through the assistance of other Bureau of Mines Metallurgy Research Centers. Emphasis was given to evaluating chalcopyrite concentrates having various amounts of accessory minerals and precious metals to determine the recovery efficiency for copper, the purity of the electrowon copper, and the potential loss of valuable minor elements. All samples were characterized by optical microscopy and X-ray diffraction. Major and minor elements plus precious metals were determined quantitatively; other trace elements were estimated by semiquantitative optical emission spectrography. The elemental compositions are summarized in table 1.
The sample labeled PC-3 consisted of chalcopyrite with approximately 5 weight-percent quartz and a trace of covellite. Sample RC-1 was principally chalcopyrite with minor amounts (3 to 5 weight-percent by visual estimates) of pyrite, covellite, and bornite plus 5 to 10 weight-percent quartz. Sample AC-3 was chalcopyrite plus minor amounts of covellite, cubanite, bornite, pyrite, and sphalerite. The AC-3 concentrate had considerably more complex intergrowth of the copper-bearing sulfides as compared with the other concentrates.
Experimental Results and Discussion
Roasting
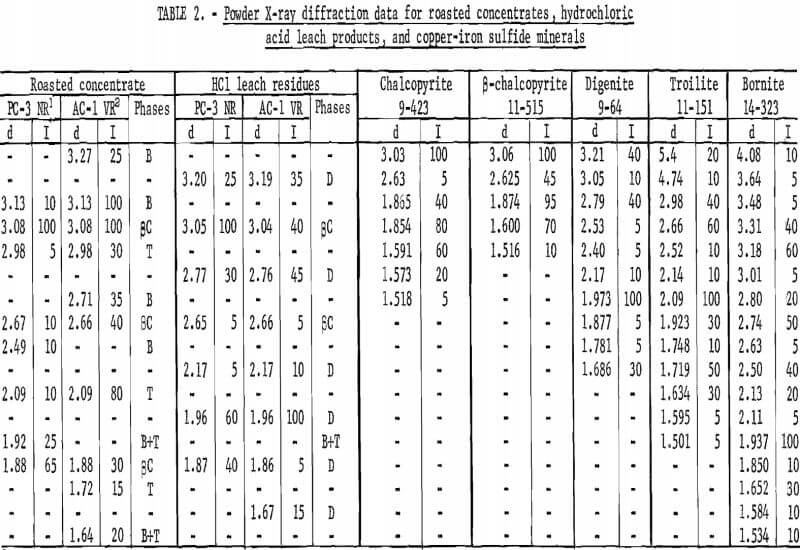
The objective of the roasting step is to convert the chalcopyrite into crystallographic phases from which most of the iron can be extracted by a hydrochloric acid leach. Experimentally, the conversion of chalcopyrite was concluded to go through an intermediate sulfur-deficient cubic chalcopyrite phase, the X-ray pattern of which is similar to β-chalcopyrite (ASTM card 11-515) but with a slightly expanded lattice. For example, the d-spacing for the 112 reflection for the observed intermediate phase was 3.08+ A as compared with 3.06 A for ASTM 11-515. This expanded lattice is easily determined experimentally as the shift from 3.04 A for natural chalcopyrite to 3.08 A corresponds to 0.4° 2θ for Cukα radiation. The expanded lattice, d112 = 3.08 A, was observed on all samples investigated; the intensities of the β-chalcopyrite lines decreased as the reaction approached completion. The solid phases in roasted samples were troilite, an unindentified high copper—low iron sulfide tentatively called a bornitelike phase, and β-chalcopyrite. Samples designated as a good roast (80 percent or higher iron extraction) gave patterns with significant peaks of troilite and the bornitelike phase. Samples whose pattern was essentially β-chalcopyrite generally gave significantly poorer iron extraction in the range from 50 to 70 percent.
For 20 percent sulfur removal during roasting, the reaction can be described by the following equation:
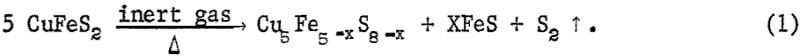
In addition, there will be variable amounts of β-chalcopyrite depending on the completeness of the reaction. The Cu5Fe5-xS8-x is considered to have a bornitelike structure because of the qualitative agreement between the authors measured data and ASTM card 14-323 . However, the Cu5 Fe5-x S8-x phase disappears during the hydrochloric acid leach of roasted samples whereas natural bornite dissolves very slowly in the same leach solution. Therefore, the Cu5 Fe5-x S8-x phase has different properties than natural bornite.
The solid residue remaining from the hydrochloric acid leach of roasted concentrate consists of digenite (Cu8S5), an altered β-chalcopyrite, gangue minerals containing the precious metals, and molybdenite. As a result of the acid leach, the β-chalcopyrite peaks decreased in intensity and also shifted to a lower unit cell. For example, the d112 spacing lowered from 3.08 A in the roasted concentrate to 3.06 A in the leach residue. The β-chalcopyrite pattern for the leach residue is in excellent agreement with ASTM card 11-515. X-ray diffraction data for a good roast, a poor roast, and their leach residues are listed in table 2 together with data for 3-chalcopyrite, troilite, digenite, and bornite.
Three roasting techniques were evaluated to determine their effectiveness in providing the required phases for efficient iron leaching. The techniques were stationary deep-bed vacuum roasting in an induction furnace, vacuum roasting in a rotary kiln, and fluidized-bed nitrogen roasting at atmospheric pressure.
Stationary deep-bed roasting of pound quantities of concentrate in a horizontal induction furnace at 850° C presented two major difficulties; only the top layer of the concentrate lost any appreciable sulfur, and ferrous sulfide produced during the roasting was pulled out of the furnace by the magnetic stirring action of’ the induction coil. The very fine ferrous sulfide passed through a liquid nitrogen trap to the vacuum pump and eventually fouled the pump oil. Therefore, this approach was discontinued.
Various concentrates were successfully vacuum roasted in a 5-inch-ID rotary kiln for 2 to 7 hours at a final sample temperature of 850° to 920° C. Samples of approximately 1 pound each were contained in zircon reactors that were rotated during the heating cycle. A vacuum of 0.1 torr was maintained with a mechanical roughing pump. Sulfur and small amounts of sulfur dioxide generated during the roasting were collected on the surface of a liquid nitrogen cold trap. Based on these studies, a roasting time of 3 hours at 920° C was selected to produce the desired phases. Although process evaluation personnel indicated that vacuum roasting would not be economically acceptable, vacuum roasting did provide adequate samples to establish the feasibility of the subsequent leaching and electrowinning steps.
The next step was to develop a method based on fluidized-bed roasting in a nitrogen atmosphere that may be economically acceptable. A 1-¾-inch-ID quartz fluidized-bed reactor was constructed for roasting concentrates with hot flowing nitrogen gas. In fluidized-bed roasting, the incoming nitrogen gas passes through 18 inches of externally heated pieces of graphite rod (pebble heated) and then passes through a porous disk fluidizing a 10- to 14-inch bed (1.3 to 1.7 pounds) of concentrate. Severe dusting was encountered when, as received, concentrate was roasted in this system. The optimum particle size range was found to be minus 8 plus 35 mesh. With the particles smaller than 35 mesh, dusting was a problem; with particles larger than 8 mesh, fluidization could not be achieved. The minus 8- plus 35-mesh size was obtained by screening the raw concentrate and by pelletizing the minus 35-mesh fraction, drying, and rescreening.
Roasting tests were conducted as follows. The charge was placed in the reactor, and a nitrogen flow rate of 11 liters per minute was started. The heaters were turned on, and when the temperature at the porous disk reached 500° C, the nitrogen flow rate was increased to 20 liters per minute. When the temperature at the disk reached 740° C, the flow was set at 26 to 29 liters per minute. A temperature profile of the reactor at this point showed 435° C—8 inches into the graphite rod heater, 620° C-12 inches into the graphite rod heater, 740° C—at the porous disk, and 800° C—13 inches above the porous disk (the top of the unfluidized charge). The concentrate was maintained at temperature for 1 to 2 hours. Subsequently, the heaters were turned off and the charge allowed to cool to room temperature with a nitrogen flow rate of 11 liters per minute.
During roasting the α-chalcopyrite was altered first to the intermediate β-chalcopyrite phase and finally to a mixture of the bornitelike phase and troilite. In a 20-minute nitrogen roast at 740° C, the calcine was found to consist of the well-crystallized β-chalcopyrite, “bornite,” and troilite. In 2-hour nitrogen roasts, more “bornite” and troilite were formed, but some β-chalcopyrite was still present in most samples.
Seven roast tests were made at 740° C for 2 hours on the three concentrates. The PC-3 concentrate roasted with little difficulty; however, the calcine consisted of principally the modified β-chalcopyrite. With the RC-1 concentrate, the bed collapsed at about 600° C (disk temperature), which prevented fluidization during the run; however, the test was continued and the crystallographic structure changes were the same as when the bed was fully fluidized. The AC-3 concentrate had a tendency to form a cinder. This same effect was observed during the vacuum rotary kiln roasting of this concentrate as cinderlike balls were formed.
Because similar structure changes were obtained with the bed in an unfluidized state, it appears that a multiple-hearth or a gravity-type furnace could be substituted for the fluidized-bed nitrogen roast. Conversion of the chalcopyrite to the bornite-type structure plus troilite is necessary to achieve maximum removal of the iron in the first-stage leach. However, total iron removal in the first-stage leach is not essential to the success of the overall technique.
During the roasting step a significant fraction, 20 to 90 percent, of the more volatile elements, such as arsenic, bismuth, lead, and zinc, are removed together with the elemental sulfur and some fine unreacted particles of the concentrate. Composition of the dust generated during the roasting step is discussed in the section “Recovery of Sulfur.”
Hydrochloric Acid Leach To Remove Iron
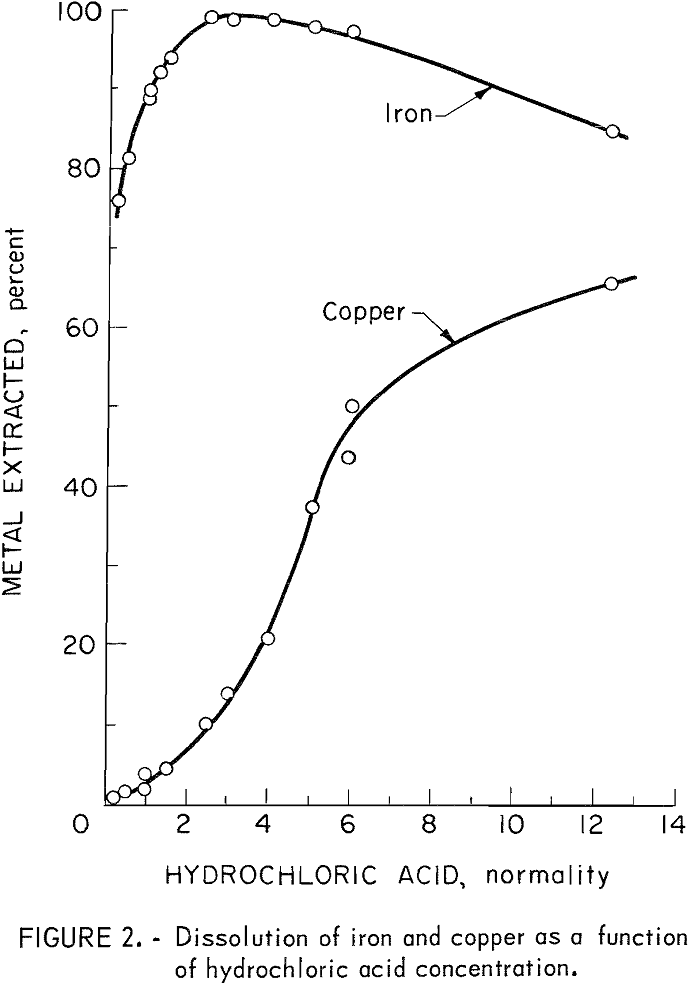
The removal of iron by a hydrochloric acid leach is based on a prior study by Tkachenke, Tseft, Dem’yanikov, and Slashchinina. They achieved 81 percent iron extraction from vacuum-decomposed chalcopyrite with a 10-minute leach using boiling 2.7 N hydrochloric acid. In the present studies, the calcines were screened to minus 60 mesh and then leached at a solid-to-liquid ratio of 10 to 16 grams per 100 milliliters of boiling hydrochloric acid solution for a leaching time of 10 minutes. The optimum hydrochloric acid strength was concluded to be 2.7 N based on the data shown in figure 2.
Up to 10 percent of the copper was dissolved during initial iron leach tests conducted for extended times. This dissolution of copper was a result of a loss of hydrogen sulfide from the leach solution. Subsequent tests confirmed that bubbling hydrogen sulfide through the solution prior to filtration would maintain the soluble copper at an insignificant level. During actual plant operation, some of the hydrogen sulfide liberated during the hydrochloric acid leach could be recycled through the leach solution to convert any dissolved copper to insoluble copper sulfide.
On studies with the vacuum-roasted samples, the iron removal was 85 to 95 percent for the PC-3 and RC-1 concentrates and in the range from 60 to 70 percent for AC-3 concentrate. With nitrogen-roasted concentrates, the iron removal was in the range from 70 to 85 percent, with the highest for the RC-1 concentrate. Without exception, roasted samples whose X-ray diffraction pattern showed moderate to major peaks of troilite and the bornitelike phase, gave good iron extractions. Tests involving longer leaching times, finer feed materials, and releaching of the residue did not remove any significant additional amounts of iron. Therefore, the key to greater iron extraction lies in better roasting rather than improved leaching techniques.
In the proposed process, the leach solution would be subjected to a hydrochloric acid regeneration step based on the oxidation of aqueous ferrous chloride with oxygen to obtain ferric oxide and hydrogen chloride. Analytical data for the various filtrates are given in table 3. The regeneration step as shown in figure 1 was not investigated in this study because available processes were considered acceptable.
Dissolution of Copper in Oxygenated Spent Electrolyte
The residue from the hydrochloric acid leach is digenite plus lesser amounts of molydenite, gangue minerals, and sulfur-deficient β-chalcopyrite. The digenite is dissolved by the spent electrolyte by the following reaction.
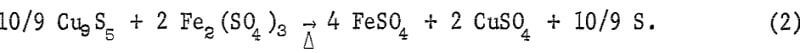
As indicated in equation 2, the ferric ion plays an essential role in the dissolution process. During the dissolution, ferric ion is reduced to ferrous ion. Therefore, oxygen is added continuously to convert the ferrous ion back to the ferric state:
![]()
The initial batch of spent electrolyte was prepared from reagent-grade cupric sulfate, ferrous sulfate, and sulfuric acid to correspond to the following concentrations in grams per liter: Copper—11.6, iron—2.9, and H2SO4—218. In the first set of tests, air was used as the oxidizing agent for converting ferrous iron to ferric during dissolution of digenite. After 26 hours at 100° C only 50 percent of the available copper was in solution, and the rate of dissolution decreased to essentially zero. Other tests using air as the oxidizing agent gave similar results. Another possible approach for converting ferrous to ferric iron is available also in a turboaerator.
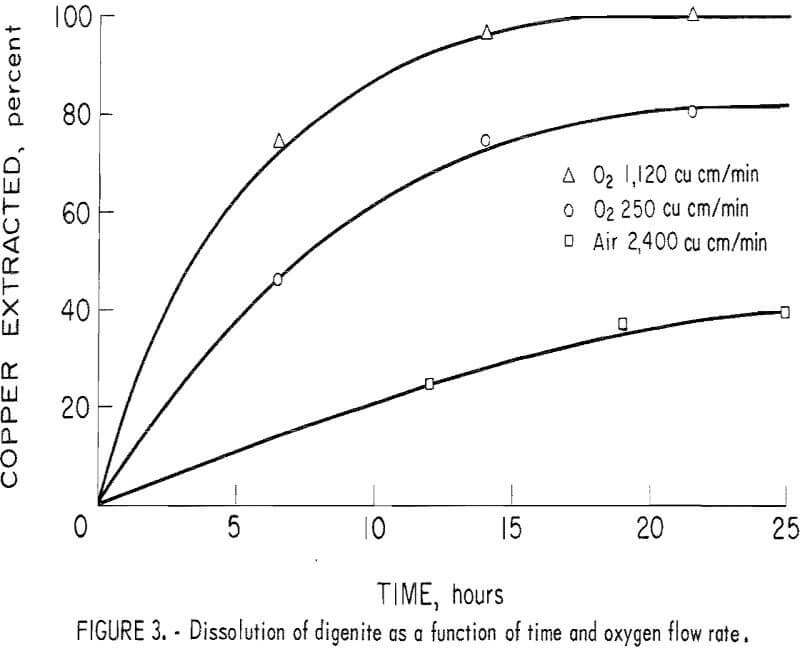
Fallwell found that four times as much oxygen could be dissolved into solution using pure oxygen as compared with using air. Comparison tests between oxygen and air were conducted at 90° C with 60 grams of first-stage leach residue and 600 ml of spent electrolyte. Agitation was provided by a 1-inch-long Teflon-coated stirring bar rotated at 435 to 535 rpm.
The oxidizing gas, air or oxygen, was bubbled into the solutions at an equivalent oxygen flow rate of 250, 500, and 1,120 cubic centimeters per minute (airflow rates of 1,200 and 2,400 cubic centimeters per minute of air are equivalent to 250 and 500 cubic centimeters per minute of oxygen, respectively). After 20 hours with air, approximately 40 percent of the copper was dissolved and the dissolution rate was essentially zero (fig. 3). In comparison, approximately 80 and 100 percent of the copper was in solution using oxygen at flow rates of 250 and 1,120 cubic centimeters per minute, respectively. In all subsequent studies, 99+ of the copper in the first-stage leach residue was dissolved after 20 to 22 hours in the spent electrolyte at an oxygen flow rate of 1,120 cubic centimeters per minute.
The spent electrolyte leaching tests were generally run serially in groups of three. Consistently, it was noted that the dissolution rate in flask 2 was
more rapid than in flasks 1 and 3 as evidenced by the earlier appearance of elemental sulfur on the solution surface. In these leaching tests , the oxygen was passed in series from flasks 1 to 2 to 3; therefore, differences in oxygen flow rate was not the reason for the higher dissolution rate noted in flask 2. Using a strobe light, the researchers found that the stirring bars in flasks 1 and 3 rotated at 435 rpm as compared with 535 rpm for flask 2, Therefore, it was concluded that the higher dissolution rate in flask 2 results from the more vigorous stirring.
An oxygen flow rate of 1,120 cubic centimeters per minute provides approximately a factor of 100 times more oxygen than is theoretically required to convert the ferrous ion to ferric. Based on the dissolution rates in flasks 1 and 3, there was no dropoff in dissolution efficiency using recycled oxygen; therefore, it should be possible to connect leaching tanks in series to the oxygen flow or recycle the oxygen through the leach solutions. Another approach is to pressure leach under an oxygen atmosphere; however, the present studies were limited to leaching at essentially atmospheric pressure.
Several tests were made to evaluate the buildup of impurities when the electrolyte is recycled after electrowinning the copper. In these tests, the electrolyte was recycled through the copper dissolution and electrowinning steps three times. Approximately 20 percent of the electrolyte was discarded after each electrowinning cycle. This discard is necessary to limit buildup of sulfate, iron and impurity elements. The increased sulfate concentration results from addition of sulfur dioxide during electrowinning to keep the iron in the ferrous state. In an operating plant, the copper in the discard would be recovered by cementation. This cement copper would be added to the electrolyte to reduce the ferric iron to ferrous thus saving sulfur dioxide in addition to making more copper available for electrowinning.
The concentration of the eight elements followed in this recycling study are listed in table 4 for the initial synthetic spent electrolyte and for the electrolyte after the first, second, and third cycle. Elements that form very insoluble sulfides, such as lead and silver, do not show any buildup because their concentration is controlled by their sulfide solubility product. Impurities such as aluminum, calcium, and magnesium increase steadily until they reach a maximum level established by the amount of solution discarded after each electrowinning step. For 20 percent discard, the maximum buildup is approximately five times the concentration of the first cycle leach solution. Iron builds up to its maximum level more rapidly because of its high starting level, 2.9 grams per liter, in the synthetic spent electrolyte as compared with essentially zero for most of the other elements. The data for iron in table 4 are representative of a sample with 80 percent of the iron removed in the hydrochloric acid leaching step.
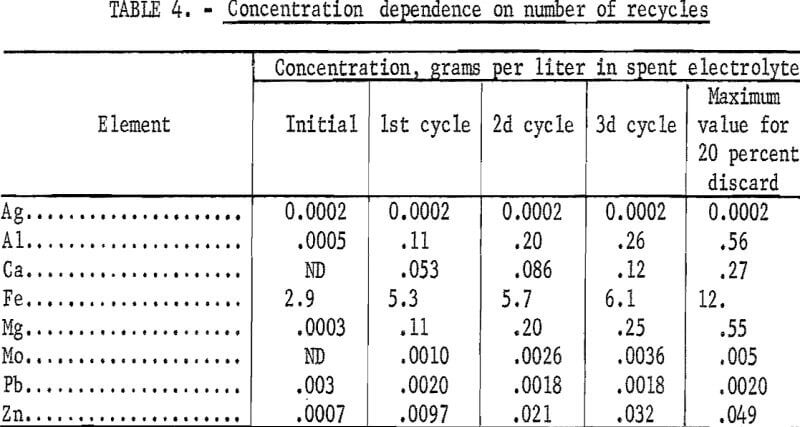
A brief study was made comparing dissolution rates for copper and iron in spent electrolyte using the raw concentrate, roasted calcine, and the hydrochloric acid leach residue. Sample sizes were selected to be equivalent to the copper contained in 60 grams of the hydrochloric acid leach residues. All samples were leached in spent electrolyte for 22 hours using the optimized conditions described earlier in this section. Only approximately 30 percent of the copper was dissolved from the raw concentrate as compared with 100 percent for the calcine and the hydrochloric acid leach residue. However, the iron concentration in the solution from the calcine was 17.8 grams per liter as compared with 4.1 grams per liter for the hydrochloric acid leach residue solution. Therefore, after five cycles of the electrolyte through the dissolution and electrowinning steps using the roasted concentrate directly, the iron concentration would be in the range of 50 to 60 grams per liter using a 20 percent discard per cycle. Thus, the first-stage hydrochloric acid leach is necessary to prevent the buildup of a prohibitively high level of iron and other metals in the electrowinning solution when starting with copper-iron sulfides.
Electrowinning Copper
High-purity copper, 99.99+ pure, was electrowon at a high cathode current efficiency and a low cell voltage from thirteen second-stage leach solutions containing the following in grams per liter: Copper—26 to 33, iron—3 to 9, and sulfate—225 to 240, plus minor amounts of sulfate soluble impurities, (table 5). A graphite anode, combined with the injection of sulfur dioxide gas into the electrolyte, was substituted for the conventional electrowinning process that employs a lead-antimony anode. The graphite anode showed no visible signs of erosion after the equivalent of 15 days of continuous operation. In the conventional lead anode process, lead contamination of the cathode deposit by erosion of the lead anode is a significant problem.
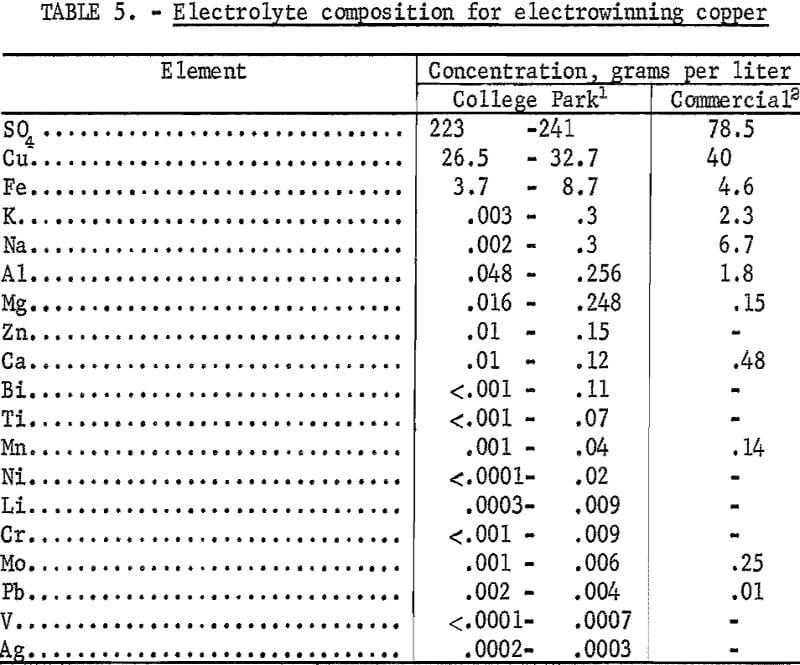
All electrowinning tests were conducted in a 2-liter glass beaker using 1.6 liters of electrolyte. The cathode was a 1-inch-diameter titanium rod having an effective plating area of 0.040 square foot. A graphite cylinder, inside diameter of 3.5 inches and a wall thickness of 1/8 inch, served as the anode. Surface area of the anode exposed to the plating solution was 23 times that of the cathode. The tests were conducted at 55° C with agitation provided by a 3/8-inch-diamcter by 2-inch-long Telfon-coated stirring bar rotated at 340 rpm. Temperature was maintained by a 250-watt infrared lamp with the on-off cycle controlled by a thermoregulator. The electrowinning cell was operated at 0.8 ampere corresponding to 20 amperes cathode current per square foot. Sulfur dioxide gas was bubbled through the solution at a rate of 10 to 50 cubic centimeters per minute. Even at the lowest flow rate, sulfur dioxide was escaping from the solution. The electrowinning tests on the various second-stage leach solutions were conducted on both an intermittent, 3 to 6 hours, and a continuous basis until the copper concentration was reduced to a level of approximately 10 grams per liter. In the intermittent tests, the cathode deposit was stripped, and the electrodeposition was restarted after each test interval.
Sulfur dioxide added during electrowinning decreases the concentration of oxygen at the anode and reduces the ferric ion to the ferrous state. Ferric ion reduces the cell efficiency by reacting with cathodic copper to form ferrous sulfate plus cupric sulfate. Oxygen liberated at the anode reacts with ferrous sulfate to give ferric sulfate thus completing the undesirable oxidation-reduction cycle at the two electrodes. Thus the following two reactions are essential to achieve the high current efficiency obtained in these tests:
SO2 + H2O + ½ O2 → H2SO4……………………………………………………………….(4)
and, Fe2(SO4)3 + SO2 + 2H2O → 2Fe2SO4 + 2H2SO4……………………………..(5)
Another factor to be considered is the low cell voltage, 0.4 to 0.5 volt, as compared with the normal electrowinning cell voltage of 1 volt or greater. This lower cell voltage results in a significant reduction of power requirements. The low voltage achieved in these tests results from eliminating the oxygen overpotential by adding sulfur dioxide as given in equation 4.
Copper samples obtained from seven of the test solutions were analyzed for trace elements by quantitative optical emission spectrography. In all of the samples, the following elements were below the limit of detection given in parts per million: As-2, Au-1, Bi-0.5, Ni-3, Sb-3, Sn-2, and Te-4. Iron and lead were present at the 5- and 10-ppm level, respectively, in one set of samples but were below the limit of detection, 4 and 2 ppm, respectively, in all other samples. Silver was within the range of 10 to 50 ppm in the first several grams of copper electrowon from any solution but was not detectable, <1 ppm, in subsequent deposits from the same solution. This preferential deposition of silver in the initial deposits is expected since silver is more easily reduced than copper. Therefore, the ratio of silver to copper in the deposit drops rapidly with electrowinning time. No other significant metallic impurities were detected by a general qualitative spectrochemical procedure. Sulfur, a common impurity in electrowon copper, was determined by a combustion titration technique. Sulfur was in the range from zero to 15 ppm in all but one of the deposits analyzed; the sulfur ranged from 15 to 40 ppm on various portions of one deposit. In general, the copper deposits meet or exceed the ASTM specifications for most classes of high-purity electrorefined copper and significantly exceed the purity of conventional electrowon copper.
Recovery of Sulfur
Two steps in the proposed process generate elemental sulfur-the inert gas roasting and the second-stage leach. The roaster offgas solids contain approximately 85 weight-percent sulfur, and the second-stage leach residue ranges from 40 to 80 weight-percent sulfur; the value for the second-stage leach residue depends on the gangue mineral content of the concentrate.
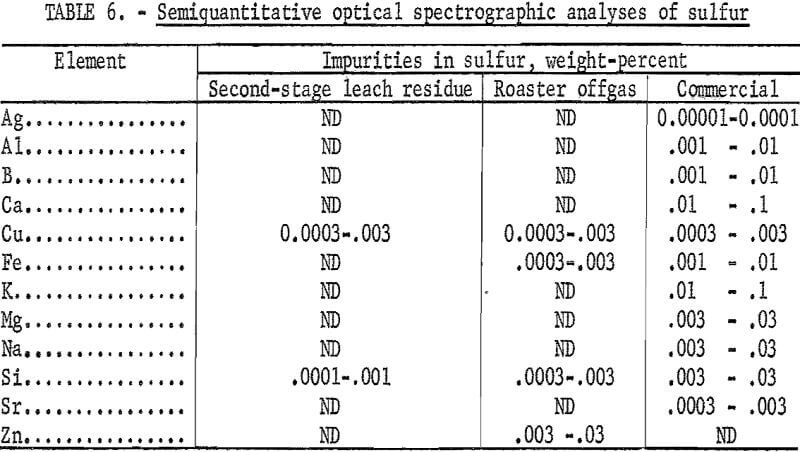
Sulfur was recovered from both sources using a method described by Haver and Wong. Their method is based on the wide variation in solubility of sulfur in perchloroethylene with temperature. At 25° C, the solubility is 25 grams per liter compared with 422 grams per liter at 100° C. The sulfur-bearing samples were slurried with boiling perchloroethylene (boiling point 121° C) to dissolve the sulfur then filtered while hot. Because of its density, perchloroethylene can be heated to boiling in an open vessel without any appreciable loss if a cooling ring is provided above the liquid level. Bright yellow crystals were obtained from the second-stage leach residue. Sulfur from the roasting offgas solids was brownish-yellow and did not significantly change with recrystallization.
Optical spectrographic analysis indicated that the recovered sulfur from both sources was of higher purity than a commercial grade of sulfur (table 6). The only significant differences in detectable impurities between sulfur from the roaster offgas solids and sulfur from the second-stage leach residue was trace amounts of iron and zinc in the offgas solids sulfur.
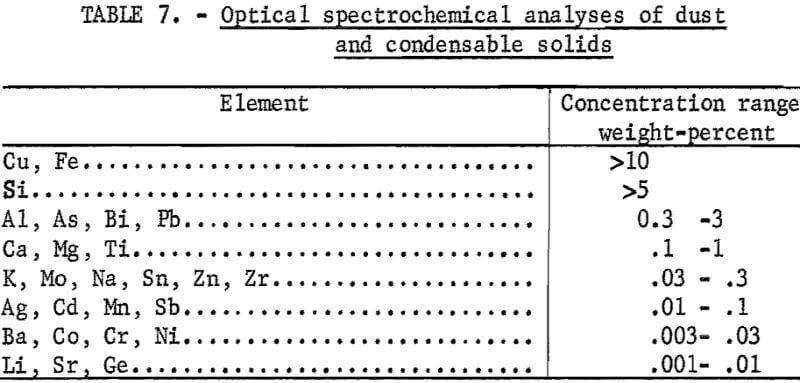
Semiquantitative spectrochemical analyses of the dust and condensable solids collected together with sulfur during the roasting step are given in table 7. These data represent the composition of the sample after extraction of sulfur. The nonsulfur fraction represents 0.5 to 1 weight-percent of the concentrate feed to the roaster, Therefore, the loss of copper due to blow over of fines is less than 0.3 percent of the copper available in the feed sample. Although the analytical data are only semiquantitative, the concentrations of volatile elements in the dust fraction are significantly higher than in the initial concentrate.
Gold, Silver, and Molybdenum
Gold, silver, and molybdenum are concentrated in the residue from the spent electrolyte leach. Following extraction of the elemental sulfur from the residue, the weights of the gangue materials plus molybdenite correspond to 3.5, 10, and 13 percent of the AC-3, PC-3, and RC-1 concentrate weights, respectively. The significantly higher gangue mineral fraction for PC-3 and RC-1 concentrates correspond to the higher silica values for these samples. Analytical data for a material balance indicate that essentially all of the molybdenum and gold plus 95 percent of the silver in the roasted samples report in the spent electrolyte leach residue. There is some indication of a small loss in silver and gold during the roasting operation; however, this may be analytical error involved in tracking quantities at the part-per-million level. X-ray diffraction studies show that the molybdenum is present as molybdenite. The mineralogical distribution of gold and silver was not determined.
As a result of the significant enrichment during the leaching steps, the spent electrolyte leach residue corresponds to a very high grade ore. The silver concentration ranges from a low value of approximately 1,000 ppm for the residues from the RC-1 and PC-3 concentrates and 17,000 ppm for the AC-3 residue. Gold is present at the 50- to 100-ppm level in the AC-3 and RC-1 residues. The gold content was not followed in processing the PC-3 concentrate because of its very low concentration. Molybdenum is present in the several weight-percent range in the spent electrolyte leach residues for all three concentrates.
No studies were conducted on the recovery of the gold, silver, and molybdenum. It should be emphasized that the value of the gold and silver in some of the chalcopyrite concentrates approximates that of the copper; therefore, efficient recovery of the precious metals should be an integral part of an economically acceptable process.
Conclusions
A combined nitrogen roast two-stage leach electrowinning method shows promise as a potential process for recovering copper, sulfur, iron oxide, and valuable minor metals (gold, silver, and molybdenum) with a minimum impact on the environment. Copper of 99.99+ purity is electrowon with an overall recovery of 98 to 99 percent. Sulfur is obtained in the elemental form from both the roasting and leaching steps. Iron is available in the hydrochloric acid leach solution for recovery. Gold, silver, and molybdenum are concentrated in a silica residue.