
Table of Contents
The Bureau of Mines has evaluated the substitution of platinum-group metal coatings for bulk platinum-group metal objects as a means of reducing the consumption of the platinum-group metals. The Bureau has conducted several studies of the electro-deposition of the platinum-group metals from molten alkali metal cyanide baths during the last two decades. The major incentive for this work has been the need to protect materials from increasingly hostile environments imposed by modern technology. To withstand high-temperature environments, structural materials must possess high-temperature strength as well as resistance to oxidation and corrosion. Refractory metals such as molybdenum, tungsten, and columbium, and alloys of these metals that have the required high-temperature strength, are readily oxidized. Protection of the refractory metals could be accomplished by coating them with a suitable platinum-group metal, and a composite material with highly desirable properties would result. Bureau research has focused on producing platinum-group metal coatings on refractory metals as well as on more common materials of construction. This research has shown that high-quality deposits which are thick, adherent, and coherent can be prepared of each of the platinum-group metals, as well as select binary alloys of these metals.
Historical Perspective
In 1937, Atkinson obtained a patent describing the use of a molten cyanide electrolyte for stripping and redepositing platinum-group metal coatings from and onto base metal substrates. Atkinson used an inert atmosphere of nitrogen over the molten bath; however, he did not describe his apparatus nor the results of his experiments in detail. Twenty years later, Withers and Ritt presented a paper on the deposition of iridium from molten cyanide electrolytes and obtained a patent on this process in 1960. The plating bath used was a 70 wt pct sodium cyanide-30 wt pct potassium cyanide electrolyte at 500° C under an argon atmosphere. Iridium was added to the melt by passing 60 Hz ac at 10 mA/cm² between two iridium electrodes. In 1962, Rhoda described the electrodeposits of several platinum-group metals obtained from either pure sodium cyanide or an eutectic mixture of 53 wt pct sodium cyanide and 47 wt pct potassium cyanide. A flow of argon gas around the crucible was used to reduce the levels of moisture and oxygen over the bath. Rhoda’s study was the most extensive early work on platinum-group metals electrodeposition from the molten salt. Coatings of iridium, platinum, and ruthenium were obtained, but he was unsuccessful in depositing palladium and rhodium.
Beginning in 1967, the Bureau conducted research on the preparation of platinum-group metal coatings. Initially, Schlain and coworkers from 1967 to 1977 investigated plating from several cyanide and cyanide-cyanate or cyanide- carbonate melts. They were successful in designing a plating system that excluded oxygen from the molten electrolytes and, based upon results obtained from these experiments, made recommendations as to the necessity of including or excluding oxygen. Schlain and coworkers were successful in depositing thick coatings of platinum, palladium, iridium, and ruthenium. Platinum and iridium objects were also electroformed using copper or molybdenum mandrels. A summary of the literature on electrodeposition from molten cyanide melts up to 1977 appears in an article by Harding.
Starting in 1976, articles were published by several workers from South Africa who used the molten cyanide salt system to separate platinum-group metals by solvent extraction. These articles have reported on the purification of the molten salts, the chemical state of the platinum-group metals in both the molten salt and the quenched salt, redox extraction from the molten salt, and the cyclic voltammetry of the platinum-group metals in the molten salt. Although these publications do not discuss deposition techniques, they are important because they contain information on the observed chemical states of the platinum-group metals within the molten cyanide system.
Harding reported attempts to produce a platinum-iridium alloy coating, but was unable to produce a sound alloy deposit of predetermined composition. The first successful method of producing a platinum-rhodium alloy coating wherein the composition could be precisely varied was devised by Flinn and Manger.
Recent Bureau studies have resolved the ambiguity of the effect of oxygen through the use of electrochemical techniques, determined the physical properties of platinum coatings, and examined the effect of current reversal and pulse plating on the prorsity of platinum coatings. In 1980 Sethi and McBurney reported on studies of platinum electrodeposition from molten cyanide melts onto nickel and nickel superalloy substrates.
Only one company is known to presently use the molten salt electrodeposition process commercially. Several publications that describe the process have appeared since 1975.
Scope of Report
This Bureau report describes the procedures involved in, and the expected results of, using molten cyanide electrolytes for electrodepositing the platinum-group metals. The methods used by the Bureau to produce the platinum-group metal coatings have changed over the years as understanding of the process has improved. This report describes the practical steps Bureau researchers have found necessary in both electrolyte and substrate surface preparation to insure that a high-quality deposit is obtained. Typical coating morphologies are also shown.
Electrolyte Preparation
Many variations on the composition and purity of molten alkali cyanides have been used, from sodium cyanide to mixtures of sodium and potassium cyanide. Recent research at the Bureau has been standardized on a 50-50 mol pct mixture, and the discussion of electrolyte preparation is focused on this mixture. The reader should, however, be aware that no evidence exists to demonstrate an advantage of any particular combination of sodium and potassium cyanide in terms of deposit quality. In general, a higher operating temperature for the plating bath permits higher plating rates because of improved mass transport, but it also increases the thermal decomposition of the plating bath, resulting in precipitation of the dissolved platinum-group metal(s).
Drying and Fusing of Salts
Reagent-grade sodium and potassium cyanides are initially dried in a vacuum furnace at 250° C for 48 h. A liquid nitrogen cold trap is used to prevent back diffusion of oil vapors from the vacuum pump. Equimolar mixtures of the dried sodium and potassium cyanides are fused in air until the melt is clear and free of suspended solids, usually requiring 1 h at 515° to 600° C. The molten salt is decanted into a fused-silica tray to produce a thin solid sheet of salt. The salt and tray are then immediately placed into a helium-filled environment chamber containing less than 1 ppm O2 and H2O.
A procedure has been reported for the purification of the alkali cyanide, involving the use of silicon to remove carbonates, that is claimed to result in virtually zero electrochemically and spectrochemically active impurities. Impurities such as carbonates and cyanates can also be removed by electrochemical oxidation. However, deposits obtained from platinum baths prepared by vacuum drying of the salts, as described, were not different in quality from deposits obtained from melts that were silicon treated or electrochemically oxidized. We therefore infer that the small, residual levels of cyanate, carbonate, and carbon impurities have little or no effect on the deposit quality. A higher purity level is required to produce a more stable electrolyte for palladium plating, although this greater purity does not entirely prevent precipitation of dissolved palladium.
Platinum-Group Metal Addition
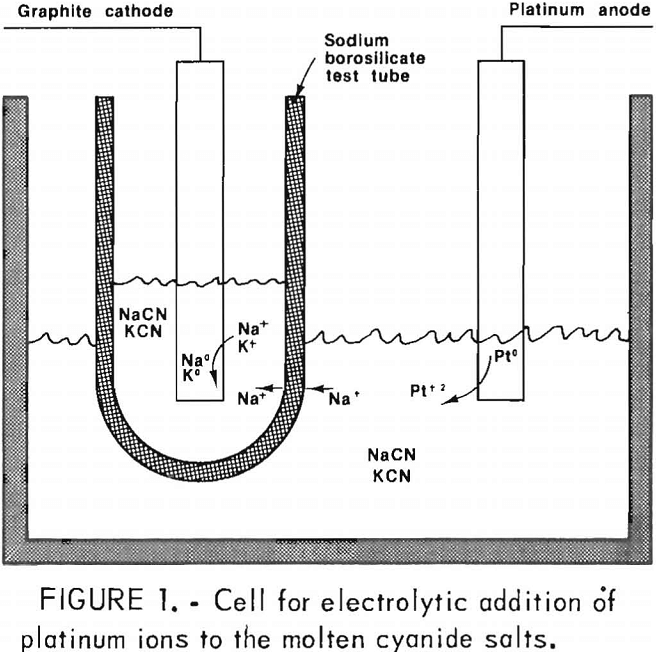
Typically, 100 to 150 g of the equimolar mixture is placed into a high-purity Al2O3 crucible and heated to the desired melt temperature, normally 570° C. All electrolyte preparations are accomplished in a helium-filled environmental chamber described in RI 8656. Platinum-group metals are added to the electrolyte by electrolytic (anodic) dissolution of the platinum-group metal of interest. The example for platinum addition is shown schematically at the bottom of the page.
As shown in figure 1, a sodium-ion-conducting membrane is used to separate the cathode and anode compartments. Oxidation products formed at the anode, Pt+², are free to diffuse into the melt, while reduction products, Na° and/or K°, are isolated within the test tube used as a cathode compartment.
In initial studies, the platinum-group metals were added to the electrolyte under potentiostatic control of the anode. The reference electrode is comprised of a small, closed silica or borosilicate glass tube filled with silver chloride containing 5 mol pct of sodium chloride. A silver wire contacts the AgCl-NaCl mixture. This tube is dipped into the molten plating bath when coatings are being electrodeposited. Although this reference electrode is not useful for thermodynamic measurements because of the

unknown differences in ion activities across the glass separator, this reference is very stable and reproducible. It was determined that, as long as the potential at the anode did not become more positive than approximately 1.1 V versus the reference electrode, cyanogen (NCCN) generation did not occur. Dissolution currents of at least 100 mA/cm² were readily attained for the platinum-group metals studied (Ir, Pd, Pt, Ru, Rh) without producing the undesirable cyanogen product.
Cathode overpotentials are of no concern during the addition of the platinum metal because that process is not limited by alkali metal concentration and the cathodic reaction products are contained in the sodium borosilicate test tube, which is discarded. The practical limitation on anodic dissolution current is the amount of undercutting that occurs on the platinum metal electrode at high current density. Because the undercutting results in a physical loss of the platinum-group metal from the anode by flaking, an anodic current density of 25 mA/cm² or less is normally used during the addition of the platinum-group metal.
With no flaking, anode dissolution current efficiencies were near 100 pct for platinum and rhodium anodes based upon the formation of Pt+² and Rh+¹. For specific information on the preparation of platinum, palladium, iridium, and rhodium plating baths, the reader is referred to RI 8656.
After the current-voltage relationships of the anodic dissolution process were understood, the desired platinum-group metal was added to the plating bath using the configuration shown in figure 1. Current density was controlled rather than the anode potential, as had initially been done. Because of power supply voltage limitations, initial values of dissolution current were 150 to 200 mA, which slowly decayed as the glass conductivity decreased because of replacement of some of the sodium with less mobile potassium in the silicate structure. Glass test tubes, which served as the sodium-ion-conducting membranes, were changed after the current fell below 80 mA. The dissolution current limit during this procedure is mostly determined by the potential drop across the glass when the current is applied.
Role of Oxygen
A major consideration in the use of the molten cyanide system is the effect of oxygen and the necessity to exclude oxygen from the atmosphere of the plating system. The cyanide ion (CN-) reacts with oxygen at temperatures exceeding 200° C and with carbon dioxide at room temperature. This requires that the molten cyanide be handled in an inert atmosphere in order to prevent decomposition of the cyanide. The major impurities formed are cyanate (OCN-), from the reaction between molten cyanide and oxygen, and carbonate, from the reaction of carbon dioxide and cyanide.
As well as directly affecting the composition of the molten cyanide bath, the presence of an oxygen-containing atmosphere affects the ability to maintain soluble platinum-group metal species in the plating bath. It was originally believed that an oxygen atmosphere was necessary in order to prepare a platinum or palladium melt. However, recent work has shown that the presence of oxygen was required only to oxidize the cathode reduction products, Na° and K°, in order to prevent these metals from reacting with the soluble platinum or palladium and precipitating it from the melt. Use of the test tube cathode compartment to isolate the reduction products during the electrolytic addition of platinum or palladium solves this problem and allows the melts to be prepared in an inert atmosphere.
Iridium, rhodium, and ruthenium melts cannot be prepared in the presence of oxygen owing to the rapid precipitation of the platinum-group metal. In contrast, platinum-containing melts were found to be still usable after more than 115 h of electrodeposition in air, although large amounts of insoluble carbonates had precipitated. In a helium atmosphere no limit to the lifetime of platinum-group-metal-containing melts has been found.
Electrodeposition
Substrate surface preparation techniques and coating deposition parameters are important factors in the preparation of protective coatings. Proper substrate surface preparation is essential for obtaining a coherent and adherent coating. Molybdenum- and iron-base alloys have been the principal substrates studied because of their desirable mechanical and high-temperature properties.
Substrate Surface Preparation
Molybdenum-Base Alloys
Although coatings have been produced on pure molybdenum substrates, the vast majority of work has focused on the alloy TZM (0.41 pct Ti, 0.10 pct Zr, bal. Mo). Substrates are ground initially to a 600-grit finish and then ultrasonically degreased with trichloroethylene. A potassium ferricyanide solution (3 wt pct K3Fe(CN)6, 0.5 wt pct NaOH, bal. H2O) is then used to etch the surface. Approximately 30 min of ultrasonic etching produces a lightly etched surface, which is rinsed with water and then 10 vol pct HCl. The desired surface has a uniform mat gray appearance with no discolored areas. If any discoloration occurs, the samples are reground and reetched. Often the 10 pct HCl does not completely remove the film formed during etching. In such a case the sample is dipped briefly into a fresh etching solution prior to rerinsing with water and the 10 pct HCl. This treatment is successful in removing the film. Etching solutions are used once and then discarded. Immediately prior to electrodeposition the samples are again ultrasonically degreased in trichloroethylene for 10 min. The extensive surface degreasing was found to be essential for producing a coherent coating.
Iron-Base Alloys
Adherent coatings have been produced on both ferritic and austenitic iron-base alloys. Sample surfaces are initially ground to a 600-grit finish. The samples are then stress-relieved for 2 h at the appropriate temperature (600° C for Fe10Cr), quenched in water, and pickled in 20 pct sulfuric acid. The final surface finish is obtained by electropolishing in 10 vol pct perchloric acid (70 pct acid)-90 vol pct glacial acetic acid until a mirror finish is produced. As with the molybdenum samples, the iron-base alloys are ultrasonically degreased in trichloroethylene for 10 min immediately prior to electrodeposition. It is not absolutely necessary that an electropolished surface be used; in fact, ground and etched surfaces are also successfully plated. If a ground and etched surface is preferred in order to reduce surface preparation time, the most important considerations are that the surface is (1) properly etched and (2) completely degreased.
Electroplating
Electrodeposition was performed with a three-electrode configuration to permit potentiostatic control of the working electrode (cathode) during deposition of the platinum-group metal. Potential-controlled electrodeposition was necessary to prevent codeposition of the alkali metals. Reduction potentials, the reference electrode, and the equipment used are described in detail elsewhere.
For materials that do not exhibit an open-circuit corrosion reaction, such as molybdenum, the sample can be placed directly into the molten salt and the deposition potential applied. However, materials such as iron-base alloys exhibit a significant corrosion reaction at open circuit and require special techniques to prevent cementation reactions from occurring. After studying the open-circuit corrosion potentials of iron-chromium alloys, it was determined that an additional 100 mV of cathodic polarization beyond the deposition potential was required to prevent cementation reactions. To prevent these reactions from occurring when the sample is initially placed Into the molten salt, the potential is applied prior to immersing the sample in the plating bath. The deposition potential can be adjusted to the desired plating potential once the sample has a coherent coating on the surface.
The high operating temperature of the molten salt plating system does not adversely affect the purity of the deposit as a result of substrate diffusion into the coating, with one notable exception. Copper or copper alloy substrates such as Monel were found to diffuse copper throughout the entire coating cross section during the plating process.
In the following sections, the operating parameters and some of the micro-structures obtained are discussed. The effects of various plating techniques on the porosity of platinum coatings are also summarized.
Platinum
The reduction potential of Pt+² (1 wt pct in the equimolar NaCN-KCN bath) to Pt° is approximately -1.7 V with respect to the Ag-AgCl reference electrode used at the Bureau. Slow (2 mV/s) cathodic polarization scans on a vitreous carbon electrode showed that the diffusion-limited current for a melt containing 1 wt pct Pt+² is between 12 and 15 mA/cm² at 580° C. The scans also exhibited an oxidation wave with a magnitude that varied from melt to melt. Although no study was made of the oxidation wave, some similarities exist between this behavior and that observed in a palladium-containing melt. This oxidation wave did not have a detrimental effect on deposit quality, regardless of its magnitude.
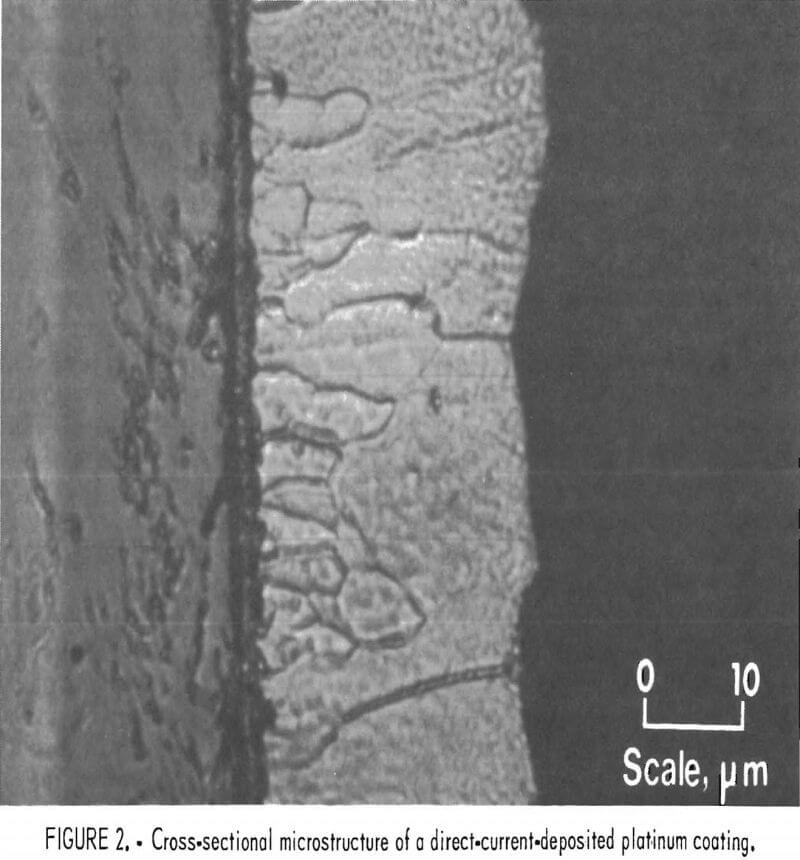
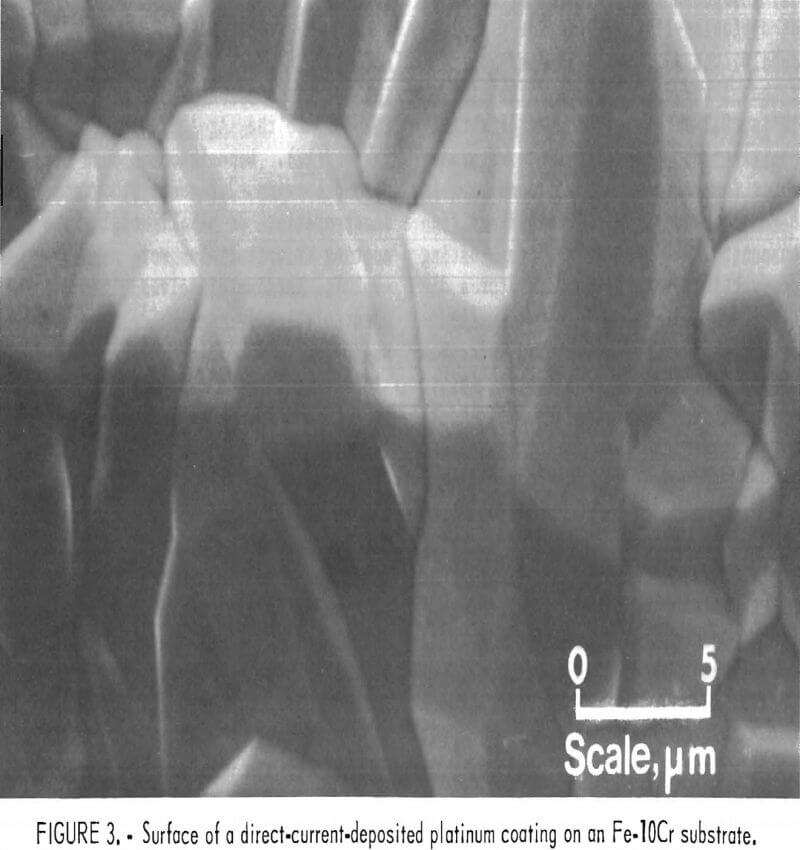
Direct current deposits were porous regardless of the plating current density. However, the least porous and best quality deposit was obtained at one-half the diffusion-limited current. At one-half the diffusion-limited current for the 1-wt-pct-Pt+² bath, the plating rate is 0.2 µm/min. Without agitation, deposition current efficiencies were near 100 pct for cathode current densities in the range of one-fifth to complete diffusion control. A typical platinum coating cross-sectional microstructure for a direct current deposit is shown in figure 2, where the average grain size increases with coating thickness. Scanning electron microscope examinations of the coating surface reveal the highly crystalline nature of the coatings (fig- 3).
Studies performed on the porosity of the platinum coatings indicate that samples plated under pulse current and current reversal (periodic reversal) electrolysis had reduced porosity. Samples that were pulse-plated using a peak current density of 25.4 mA/cm², an “on” time of 0.1 ms, and an “off” time of 0.67 ms produced the minimum porosity coating.
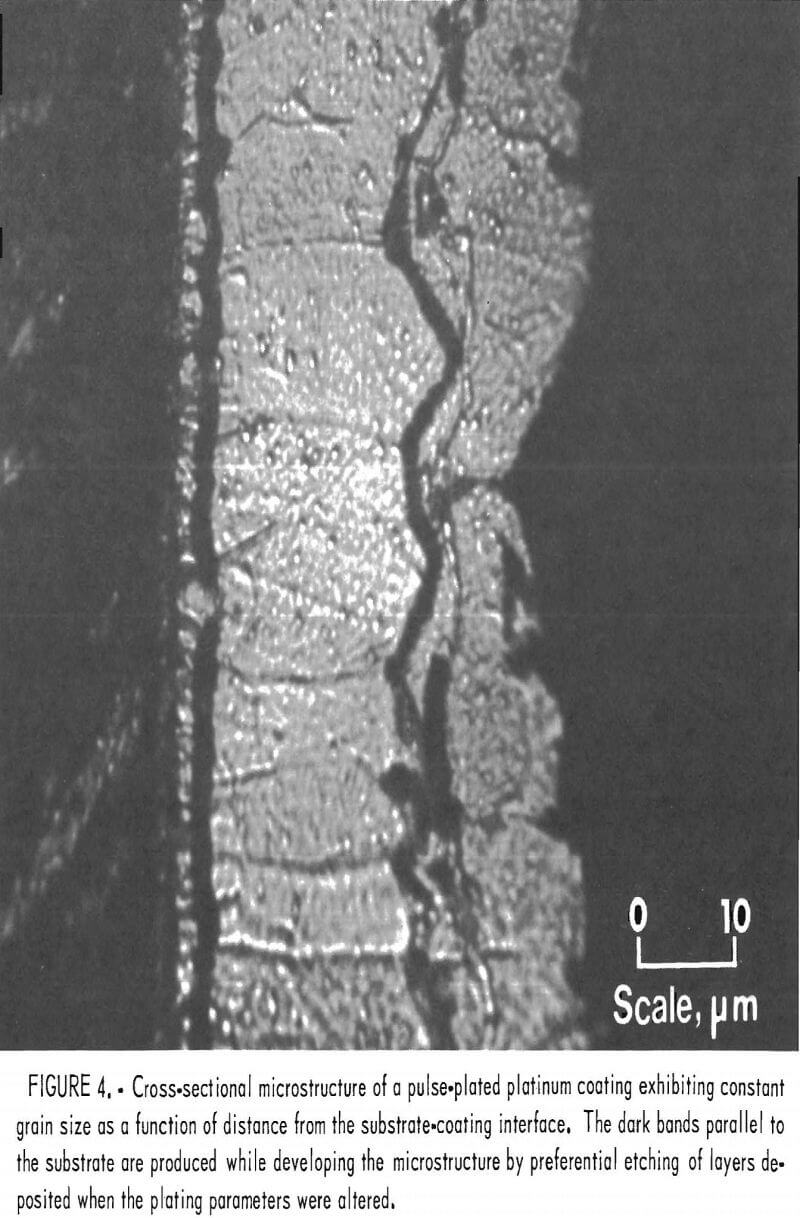
A combination of pulse plating and current reversal could perhaps produce a truly pore-free coating; however, this was not investigated. Pulse plating also produced an almost constant grain size microstructure (fig. 4). The constant grain size region, approximately 17 µm in width, is visible between the dark bands. The dark bands parallel to the substrate are produced while developing the micro-structure by preferential etching of layers deposited when the plating parameters were altered. For a more detailed discussion of the banding effect see RI 8829. The major disadvantage to the use of pulse plating was that the average current density was only 3.3 mA/cm², resulting in plating rates of only 0.09 µm/min.
Physical properties of the direct-current platinum deposits have been measured. The measured fracture or adhesion strength was 331 MPa (48,000 lb/in²), and the cross-sectional microhardness, which was found to be dependent on microstructure, exhibited a minimum value of HK25 = 52 kg/mm².
Rhodium
Rhodium reduction occurs at -2.0 V with respect to the Ag-AgCl reference electrode. Deposition should be performed between -2.2 and -2.0 V to avoid incorporation of alkali metal into the coating at potentials more negative than -2.2 V. Rhodium dissolves into the melt electrolytically as Rh+¹. Overpotentials on the anode should be minimized by increasing the anode-to-cathode surface area ratio to prevent reduction in current efficiency due to the formation of Rh+³. Diffusion currents for a 1-wt-pct-Rh melt are approximately the same as those obtained for a platinum-containing melt. Cathode current efficiencies for deposition without use of agitation are near 100 pct. Plating rates, surface topography, and structure are also equivalent to those obtained for the platinum electrodeposits. No measurements were made on the physical properties of the rhodium coatings.
Platinum-Rhodium Alloys
When the plating bath contains both platinum and rhodium, an alloy can be deposited that contains platinum and rhodium in a weight ratio equivalent to that of the metals in the molten salt. Deposition potentials must exceed both the platinum and rhodium reduction potentials in order to incorporate rhodium into the coating. A potential of -2.2 V was used in producing the alloy coatings. Although it may be possible to vary the coating composition by reducing the plating potential and, hence, the relative rate of rhodium deposition, no attempt was made to perform these experiments. All alloy coatings were produced by adjusting the weight percent of each metal ion in the molten salt. This was accomplished by alternating the anode materials during addition of the platinum-group metal and dissolving the appropriate amount of each. Again, plating rates, surface topography, and structure were equivalent to those obtained for platinum electrodeposits at the diffusion-limited current.
Palladium
The diffusion-limited current for a 1-wt-pct-Pd melt is approximately 20 mA/ cm². Figure 5 gives an example of a palladium coating on an Inconel substrate. No measurements have been made on the physical properties of palladium deposits.
Palladium-containing melts were the most difficult to produce and maintain. The problem appears to be related to the reduction potential of palladium, -1.35 V. This potential is near the redox potential for the cyanamide (NCN²-)- dicyanamide (N(CN)2-) couple:
CN- + NCN²- = N(CN)2- + 2e-…………………………………………………………….(1)
The cyanamide and dicyanamide ions can be produced in the molten cyanide electrolyte either by electrolytic oxidation or by contacting the melt with oxygen for extended periods. Cyanamide ion has been shown to be capable of reducing Pd²+ to Pd°, and the dicyanamide ion was shown to cause rapid dissolution of metallic palladium in the molten cyanide electrolyte. Regardless of the problem of maintaining the palladium in the molten salt in a form that is electrochemically reducible to a coating, satisfactory coatings can be deposited. In preparing a melt for palladium addition, extreme care must be used in purifying the molten salts, and no melting should be done in an oxygen-containing atmosphere. Melts should be preelectrolyzed prior to

palladium additions. Further purification by the methods of Lessing is also recommended.
Iridium and Ruthenium
Iridium reduction occurs at -1.6A V with a diffusion-limited current of 15 mA/cm² for a 1-wt-pct melt. Current efficiencies were found to be dependent on anode current densities, with a maximum of 80 pct for the cathodic current efficiency. The variability in current efficiencies is due to the existence of several oxidation states of iridium within the melt. Depending on the anode current density, the iridium can dissolve in the +3 or the +1 state; also, reactions at the anode could possibly oxidize the +3 state to higher oxidation states. The hardness of the iridium coatings has been reported as HK25 = 800 kg/mm² and HK100 = 727 kg/mm². Iridium deposits are extremely fine grained (fig. 6).
Deposits of ruthenium have been obtained from the molten cyanide electrolyte; however, no electrochemical characterizations have been performed at the Bureau. Recent work in South Africa has shown voltammograms for ruthenium. The hardness of ruthenium coatings is HK25 = 610 to 935 kg/mm² according to Rhoda.

Substrate Effects
Coatings must be deposited in a uniform manner over the substrate in order to obtain the “best” deposit in terms of adherence and coherence. If the substrate has been improperly prepared, the resulting coating will have characteristic deficiencies that can be identified. The structure of the substrate also affects the type of coating obtained. Examples of the effect of substrate structure and improper surface preparation of the coating are presented below. Although these examples should help reduce the time spent in isolating problems of poor coating structure or performance, these are by no means the only types of problems that can develop in electrodepositing the platinum-group metals.
Incomplete Preparation
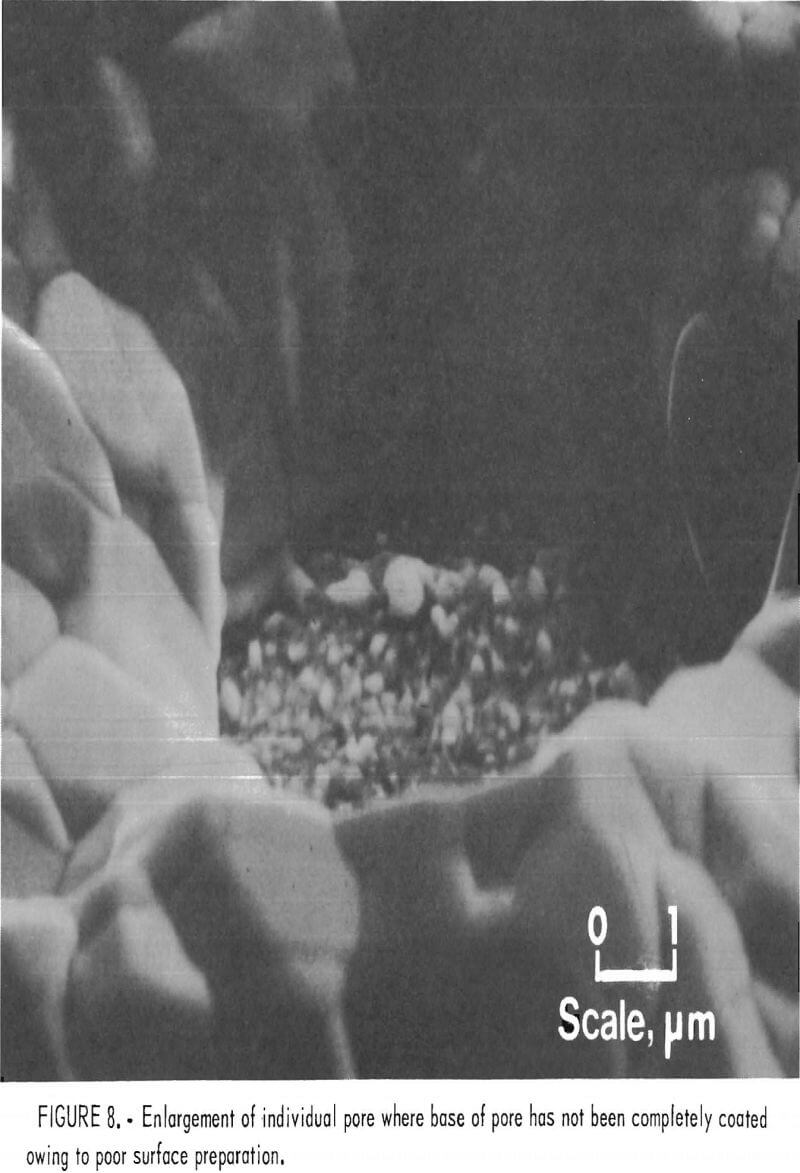
In addition to the obvious need to remove dirt and thick oxide scales from the substrates, degreasing is also a very important step in substrate surface preparation. When the surface is not completely clean, the platinum-group metal can be inhibited from depositing the continuous initial layer. The coating eventually will bridge the area if it is relatively small, but a pore or hole will persist if the area is large. Neither of these effects is desirable since they result in loss of both adherence and coherence. Figure 7 shows a sample that

has several areas that were not completely degreased. A closeup view (fig. 8) of one of the pores produced by the incomplete cleaning shows that no coating has deposited on the substrate at the base of the pore. Figure 8 also shows the columnar structure of the deposits discussed earlier.
Microstructure
The type of substrate and its structure can affect the coating morphology. Platinum electrodeposits prepared on a fully annealed iron substrate exhibit patterns similar to the grain structure of the substrate, as shown in figure 9. Thick deposits prepared on cold-rolled or annealed substrates have the same morphology, but it is only in thin deposits that the grain pattern of the substrate is mirrored. Whenever the substrate reduces the ability of the coating to nucleate, because of incomplete cleaning or type of substrate, the coating will form at the least resistant site and grow. An extreme example of this is shown in figure 10 for a carbon-carbon composite material that was plated with platinum.


Summary
The molten cyanide system is a practical medium for preparing coatings of the platinum-group metals. Coatings of high quality can be produced if care is taken in all steps of the process from electrolyte preparation to sample surface preparation to the actual electrodeposition. The single problem that remains to be solved is the obtainment of zero porosity in the coatings. Use of novel current waveforms, such as combinations of pulse and current reversal plating techniques, could result in coatings having zero porosity.
Currently, it appears that only one company has used the molten cyanide electrolyte to prepare coatings of the platinum-group metals on a commercial scale and that only platinum coatings are currently available. To the best of our knowledge, no company is using the molten cyanide system to produce electrodeposits of all the platinum-group metals. In the future perhaps the molten cyanide system can be used for the reclamation of the platinum-group metals from scrap in much the same fashion that it is currently used in South Africa to separate platinum-group metals by solvent extraction.