
Table of Contents
This report presents the results of research aimed at evaluating and preventing the leaching action that may result from the disposal of waste ferrous slags into landfills where they can be subjected to acid precipitation. Waste slag constitutes a large potential source of chromium contamination of ground waters, particularly under the enhanced leaching action of acid precipitation on waste slag dumps. Numerous investigations into the effect of acid precipitation on vegetation, surface waters, animal life, and stone architecture are to be found in the literature. The potential effects of acid precipitation on waste dump sites have not been addressed. Many sites, such as those from waste ferrous slag disposal, are not considered hazardous under normal conditions. However, the prevalence of abnormal acid precipitation conditions could lead to the possible leaching of hazardous elements from these normally innocuous wastes.
Of the numerous toxic elements that could be leached from waste slag, chromium appears to be of major concern to human health. Chromium in the hexavalent (VI) oxidation state is toxic, and several of the chromates have been deter¬mined to be carcinogenic. The U.S. Environmental Protection Agency (EPA) National Interim Primary Drinking Water Standards (proposed in 1976) restrict chromium to a maximum allowable concentration of 0.05 mg/L. Therefore, even moderate leaching of chromium from waste slag landfills into ground water supplies would have the potential of exceeding this low permissible level.
The EPA in 1979 reported the results of laboratory and field tests for various iron and steelmaking slags. They determined the slags from the basic oxygen furnace (BOF) process, open hearth, and electric arc furnace to be generally of more environmental concern than blast furnace slag. Laboratory tests showed that heavy metal components in the slags leached more readily as the acidity of the water in contact with the slags was increased. Analysis of ground water obtained from areas near iron and steel waste landfills revealed significant amounts of chromium in concentrations roughly equivalent to, or in some samples exceeding, drinking water standards.
Over 95 pct of the wrought stainless steel and heat-resisting chrome alloys produced in the United States are melted and partially refined in electric arc furnaces, followed by pouring into an argon-oxygen decarburization (AOD) vessel, where final refining and chemical adjustment take place. Slag wastes are also generated in the production of superalloy and other special chrome alloys and in the production of ferrochrome and ferrochromesilicon. The latter two products are ferroalloys that are intermediate metals in the production of chrome alloys and stainless steels. Also tested in the present work are slags from recycling operations that produce stainless steel pig metal from pelletized mill scale, grinding swarf, and other wastes from stainless steelmaking operations.
There has been considerable concern in the United States about the economic and strategic importance of chromium lost as waste in metallurgical processes. A number of Government surveys have sought to estimate the magnitude of the loss. In 1978, the National Materials Advisory Board estimated that about 30,000 tons of chromium are lost annually in slag generated with stainless steel produced by conventional methods. Cuthbertson and Ponzer reported that 77,500 tons of chromium were lost as oxides in stainless steel production in the United States during 1976. The percentage of the chromium oxides reporting to the slag was not indicated. They also reported a smelting loss of 19,815 tons of chromium during ferrochrome production in 1970. They estimated 60 pct of this chromium loss reported to the slag as oxide.
The U.S. General Accounting Office (GAO) estimated in a report to Congress in 1980 that about 21.5 million tons of steelmaking waste slag is generated each year from BOF and electric furnace operations. The GAO estimated this waste contains about 40,000 tons of chromium. If this loss of chromium is added to their estimate of chromium lost in copper smelting slags, then the total chromium disposed as waste is more than doubled, or about 98,000 tons of chromium. Most of these slags can be expected to enter into the environment as waste landfill or as construction fill and, therefore, to be exposed to acid precipitation leaching. Hence, the environmental damage caused by the leaching of chromium from these sources could be extensive.
Very little has been reported in the literature regarding the prevention of chromium leaching from waste slag. A Japanese patent claims to reduce the chromium leachability of chrome ore slag by mixing silica with the powdered slag, followed by calcining at 900° to 1,300° C„ Eguchi and Uchida studied the fixing of chromium in Cr2O3- FeO-CaO-SiO2 melts. They found the chromium leachability increased with increasing lime content and oxygen pressure but decreased with temperature. Gemmel investigated the pollution of land by a chromate smelter and found that the toxicity of calcium chromate and high pH in the soil prevented attempts at revegetation. He also concluded that dilution of the smelter waste with inert media, such as silica sand, was insufficient to allow revegetation, but that removal of the chromium from the smelter waste by chemical means was required before the waste was disposed of.
This report describes the initial laboratory research on the leachability of chromium from a variety of industrial slags from stainless steel and chrome alloy operations. The slags tested contained from less than 1 to 8.1 pct Cr. The work attempts (1) to assess the potential for the leaching of chromium from waste slag exposed to acid precipitation and (2) to recommend methods for preventing leaching through slag treatment designed to convert oxidizable, soluble chromium compounds in slags to nonsoluble forms resistant to acid precipitation.
Experimental Procedure
The slags were obtained from industrial stainless steel producers and from a chromium waste recycling operation producing pig metal for stainless steel production. The chemical analyses of the slags as supplied (base slags A, B, C, and D) are given in table 1. Base slags A and B were obtained from a stainless steel operation using the currently most prevalent stainless steelmaking technology, i.e., initial melting of pig metal and chromium scrap in an electric arc furnace (slag A) followed by the AOD process (slag B). According to the supplier, the steel being processed at the time was a 304 stainless with slag samples taken with an iron spoon and poured into an iron mold. Slag C was obtained from a recycling operation producing stainless steel pig metal from stainless steelmaking wastes. This producer tailors the slag to a basicity of 1.3 to 1.7 with appropriate additions to optimize chrome reduction and to preserve the magnesite furnace lining. The latter consideration probably accounts for the high MgO content of this slag. Slag D was (as described by the supplier) obtained from an electric furnace melt of a 300 series stainless steel. Slag D contained considerable entrained metallic particles. The compositions of base slags A, B, and C were altered for most tests by adding reagent-grade CaO, SiO2, or MgO, followed by melting in a platinum-rhodium alloy crucible to form a homogeneous solution. (These slags are designated in table 1 as A1, A6, B1 through B3, and C1 through C10.) The base slags A, B, and C were also calcined (910° C for 16 h) prior to composition alteration to oxidize any metallic particles that would attack the platinum-rhodium melting crucible. When reagents were added to base slags, the resulting slag mixtures were melted and held for 16 h above the fusion point at 1,450° C in an air atmosphere. The slag temperature was then raised to 1,500° C, and the slag was poured onto a clean, cold steel plate. In many cases, CaF2 and Fe2O3 were added to aid the pouring of the viscous slag melts from the platinum-rhodium alloy melting crucibles.
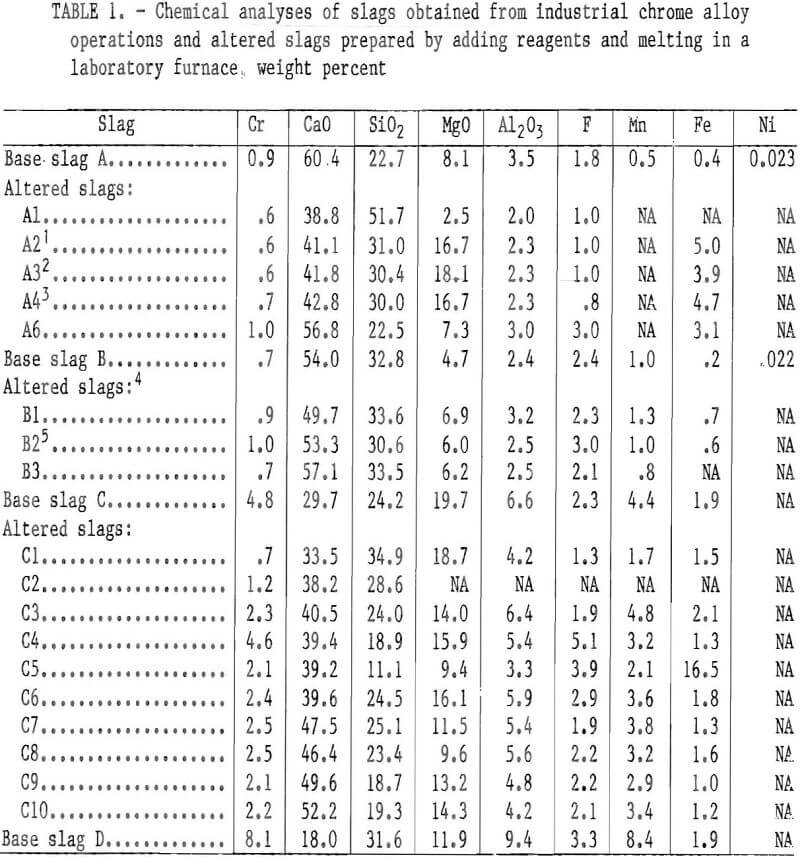
Altered slags were also prepared by adding the mineral olivine or taconite tailings plus olivine or reagent-grade MgO to the base slag A (slags A2, A3, and A4 in table 1). The olivine, a magnesium iron silicate mineral, was mined in North Carolina, according to information provided by the commercial mineral supplier. The mineral sample analyzed 52.3 pct MgO, 38.9 pct SiO2, and 7.0 pct total iron. The sample of taconite tailings came from the Minnesota Mesabi Iron Range and analyzed 64.9 pct SiO2, 16.6 pct total iron, 2.6 pct MgO, 1.7 pct CaO, 4.5 pct CO2, and 5.7 pct loss on ignition. All slags were ground to minus 100 mesh for the leaching tests. Care was taken to avoid overgrinding so as to minimize the formation of fines and. the variation of particle size between samples. The analyses for the altered slags prepared as above, with a few exceptions, are given in table 1.
Two methods were used to determine the chromium leachability of the specimen slags. The first was a long-term (7-day) test using a lysimeter-type apparatus. The second method was a simple, short- term (5- or 24-h) batch test using stirred slurries.
The lysimeter method used the apparatus depicted in figure 1 and diagramed schematically in figure 2. The leachate solution was continuously circulated through the apparatus so as to percolate through the bed of minus 100-mesh slag. The pH was measured in the upper reservoir and controlled to a selected constant value of 4.5 by automated addition of doser acid. The doser acid was usually a mixture of 0.5N in H2SO4 and 0.34N in HNO3, although the acid strength was changed on occasion depending on the slag basicity.

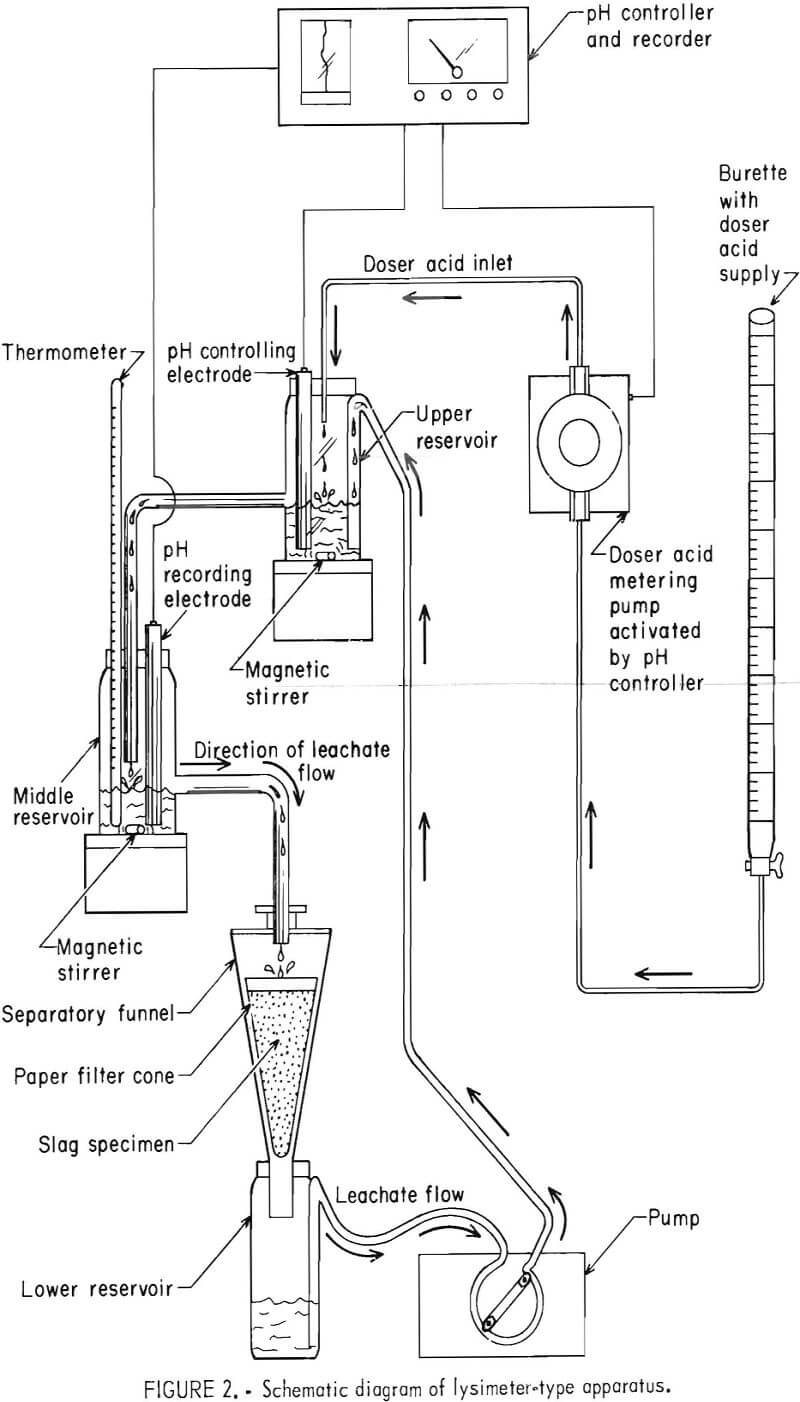
Each lysimeter experiment was begun by adding 100 g of slag sample to a cone of filter paper in the separatory funnel of the apparatus shown in figure 2. Distilled water (250 mL) was added to the lower reservoir, and the peristaltic pump was started to give a solution circulation rate of 13.0 mL/min. Doser acid immediately dripped into the system to automatically control the pH at 4.5 in response to the controlling electrode of the upper reservoir. The leachate circulation rate of 13.0 mL/min enabled a shallow pool of liquid leachate to be maintained above the slag sample, thereby providing a more uniform percolation through the slag bed. The middle reservoir recording electrode monitored the pH of the circulating leachate just before it descended to the slag sample. The 100-g slag specimen was leached 7 days (at about a 2.5:1 liquid-to-solid ratio). The leachate generated was analyzed for total chromium using standard atomic absorption spectrometric procedures.
The lysimeter apparatus was constructed almost entirely of plastic components (polyethylene reservoirs, polyvinyl tubing, and polymethylpentene funnel). A pH controller, which maintained a preset pH value to within ±0.1 pH unit, was used in combination with a positive displacement acid-metering pump (0- to 0.6-mL/min capacity) to control the pH.
In the slurry test, 10 g of minus 100- mesh slag was stirred for 5 or 24 h in 1 L of dilute H2SO4 leachant (100:1 liquid to solid). Usually, the four leachant solutions contained 3.5, 8.8, 17.6, and 35.2 g/L H2SO4. At the conclusion of the leaching period, the liquid leachate samples were collected and filtered for analyses.
Experimental Results
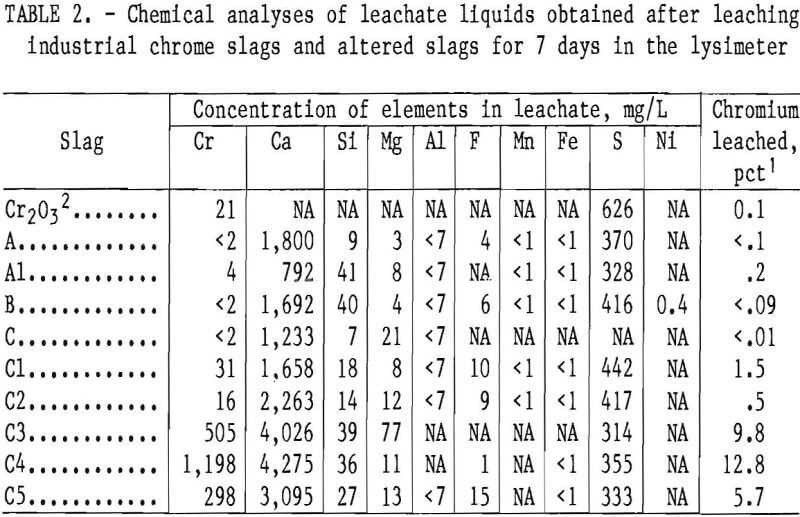
Base slags A, B, and C and several altered slags (A1 and C1 through C5) of table 1 were leached for 7 days using the lysimeter apparatus to simulate environmental leaching by acid precipitation. The simulated acid rain solution was controlled at a constant 4.5 pH using a doser acid containing H2SO4 and HNO3 in a 1.16:1 weight ratio. Also, for comparison, a physical mixture containing 4 pct Cr (as Cr2O3) in inert polystyrene powder (200 to 400 mesh) was leached under the same conditions. The leachates obtained at the end of the 7-day period were analyzed, and the results are reported in table 2.
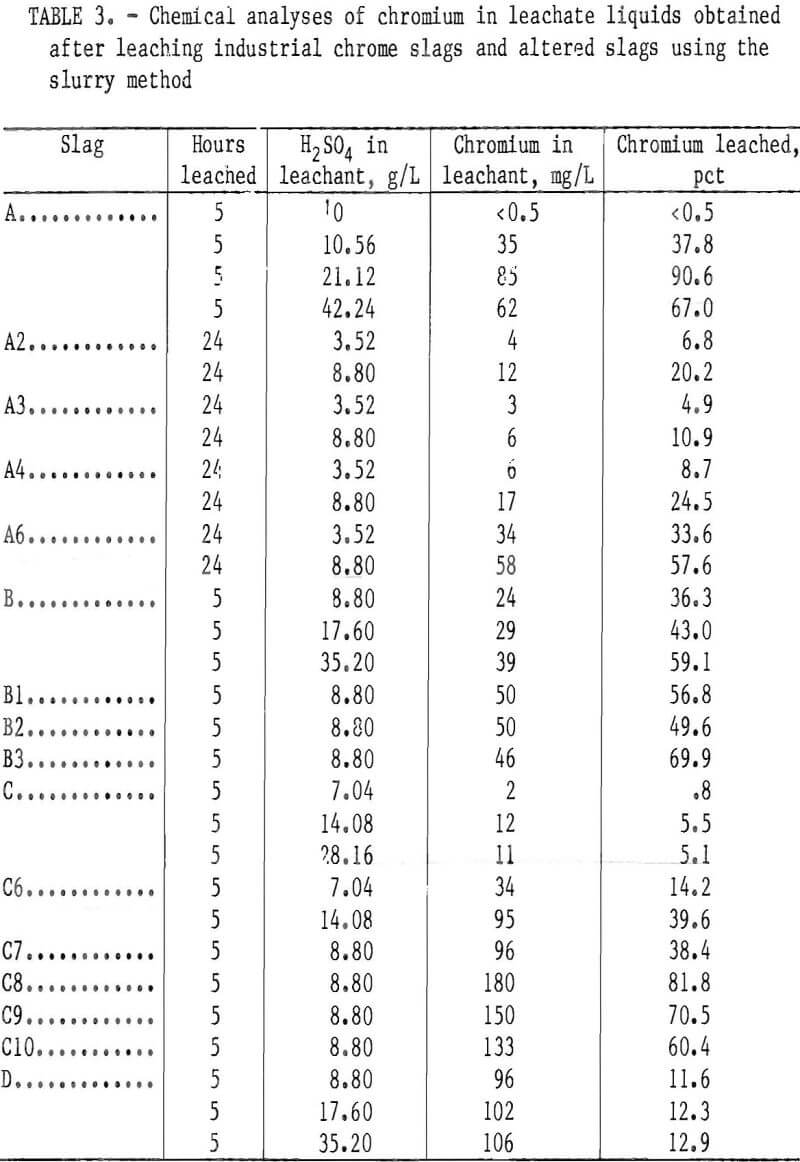
The base slags A, B, C, and D and several altered slags (A2 through A6, B1 through B3, and C6 through C10) of table 1 were leached for 5 or 24 h using the slurry method. The chromium leachability results for several leachant H2SO4 concentrations are given in table 3.
Discussion
Leaching Methods
The EPA method for determining the toxicity of solid waste has several elements in common with the lysimeter and slurry tests used in this work. However, no effort was made to duplicate the EPA extraction procedure. The EPA test has little relevancy when applied to waste ferrous slags. For example, the use of acetic acid to control the pH in the EPA test is not applicable practically or logically to waste slags. Many waste slags contain large amounts of CaO, which consume excessive volumes of a weak acid such as acetic acid to maintain the required pH level. The EPA test is modeled on the codisposal of toxic wastes in an actively decomposing municipal landfill that overlies a ground water aquifer.
Slag dumps are inorganic, relatively sterile wastes compared with the biologically active municipal landfills for which the EPA test was devised. Codisposal of ferrous waste slag with organic matter is an unlikely occurrence.
The waste slag tests in this work are based on a leachant composed of the major components of acid precipitation, i.e., H2SO4 and HNO3. In the lysimeter tests, these acids were mixed in a weight ratio of H2SO4 to HNO3 of 1.16:1, a value reported in the literature for a sampling of acid precipitation in the Eastern United States in 1975. This ratio varies throughout the United States, but the 1.16 value was considered sufficiently typical for simulating acid precipitation percolating through a waste slag heap in the northeastern, industrial area of the United States. The control pH of 4.5 was selected for Its intermediate position between acid precipitation extremes and for its ease of control with the doser acid control equipment.
Leaching of Base Slags
The lysimeter tests of base slags A, B, and C showed these slags to have good resistance to chromium leachability under the test conditions (table 2). The lysimeter results demonstrate that the solubility of chromium from these slags is less than that from pure Cr2O3, when the slags are freshly discharged from the steelmaking furnace and before they are subjected to weathering or other environmental conditions.
Table 2 also gives the leachate analysis results for a number of other elements present in the slags. The large solubilization of calcium from the slags is noteworthy, and the sulfur in the leachate originates primarily from the doser acid. The sulfur analysis is not consistent with the amount of doser acid added, because of gypsum (CaSO4·2H2O) precipitation from the leachate. Gypsum was deposited as a white powder in various regions of the leaching apparatus, particularly on top of the slag bed, and was identified by X-ray diffraction.
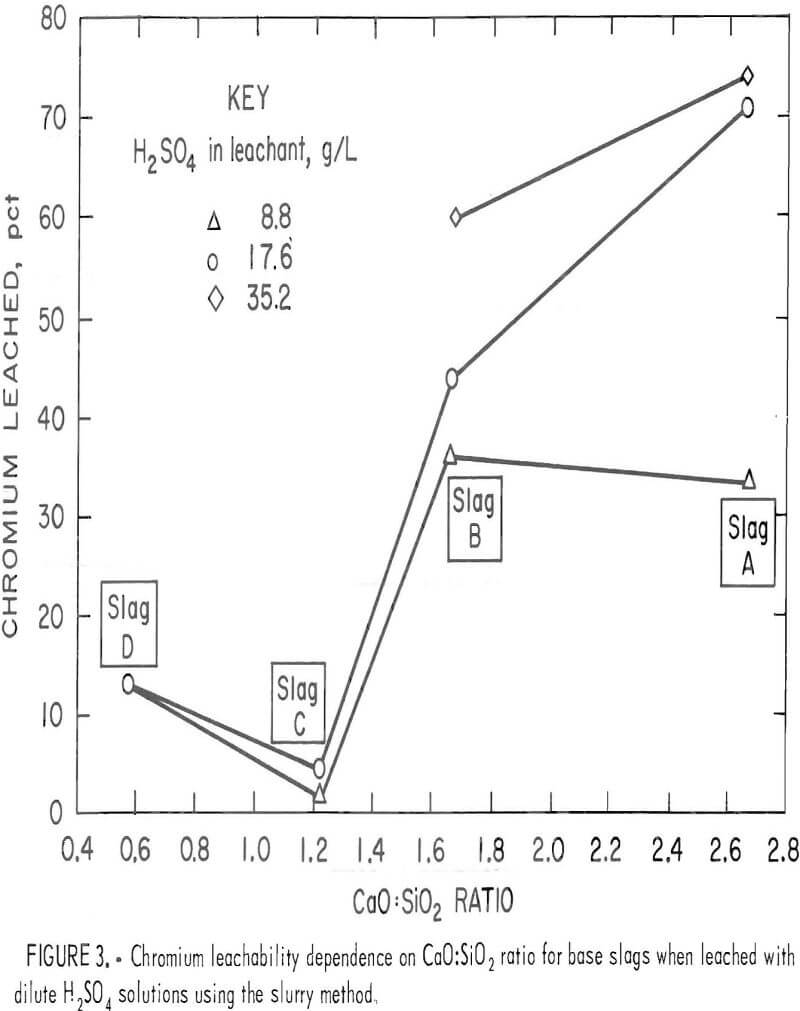
Although the chromium leachability differences between as-supplied slags were not detectable in the lysimeter testing, the slurry testing revealed marked differences. Under the conditions of slurry testing with H2SO4 leachant, base slags A and B showed considerable vulnerability toward chromium leaching, base slag D showed moderate leachability, while base slag C demonstrated high resistance to the H2SO4 solutions (table 3). The leaching susceptibility of base slags A and B in the slurry testing appeared to be influenced by the slag CaO:SiO2 ratio (the components of the ratio, CaO and SiO2, being in concentration units of weight percent). Figure 3 plots the ratio of CaO:SiO2 versus the percentage of chromium extracted during a 5-h period of slurry leaching at various concentrations of H2SO4 leachant. The percents of chromium leached from slags A and C in the figure are approximate values calculated by interpolating between experimental results in table 3 for adjacent leachants of different H2SO4 concentrations. The figure reveals a minimum chromium leachability occurring somewhere between a 0.57 and a 1.65 CaO:SiO2 ratio for all concentrations of leachant acid. For the weakest concentration, 8.8 g/L H2SO4, a maximum chromium leachability appeared to be between 1.65 and 2.66 CaO:SiO2 ratio.
Leaching of Altered Slags
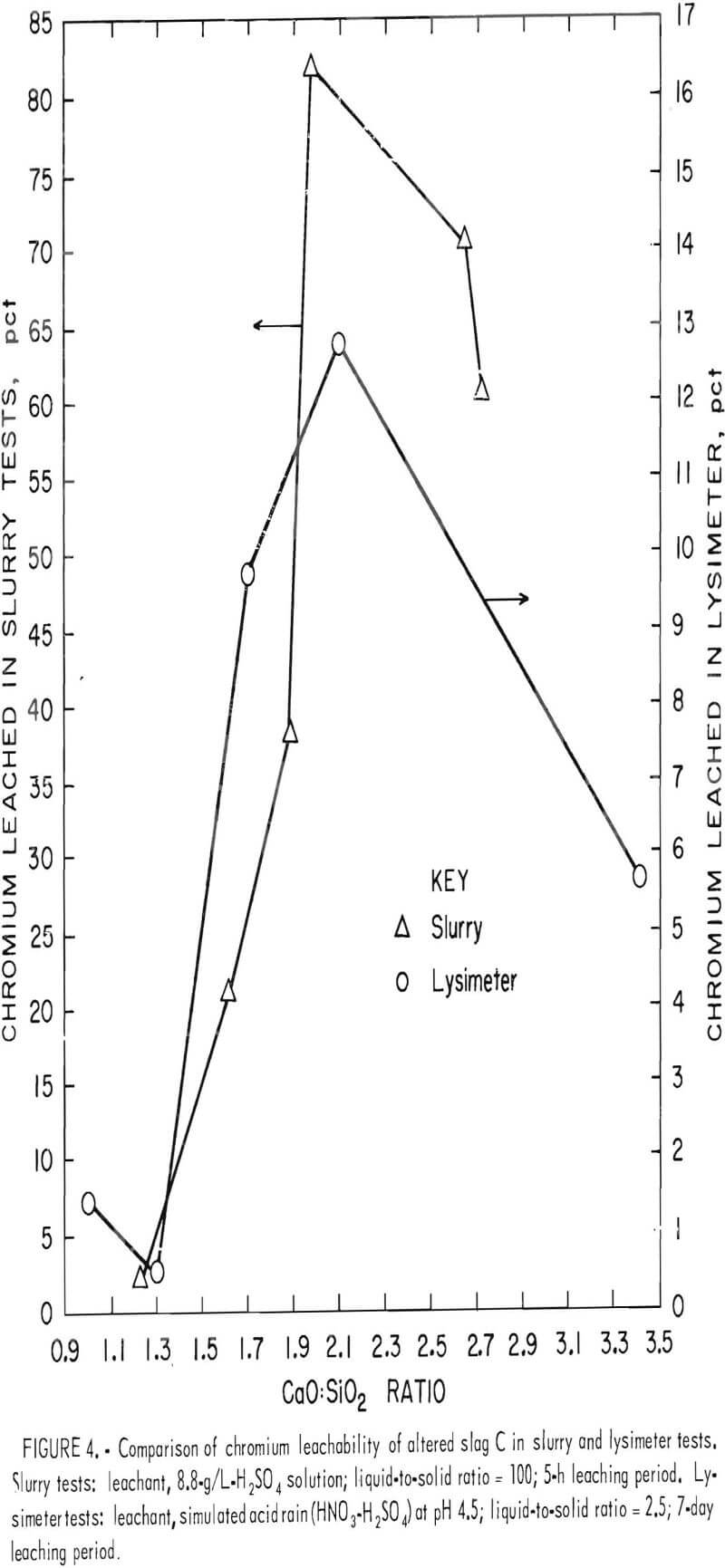
This pattern of chromium leachability dependence on the CaO:SiO2 ratio was duplicated in both the lysimeter and slurry tests when base slags A, B, and C were altered and melted in air. The chromium leachability data from the lysimeter and slurry testing for alterations of slag C (slags C1 through C5, table 2, and slags C6 through C10, table 3) were plotted against CaO:SiO2 to give figure 4. In this case the maximum leachability is more pronounced and occurs near the 2.0 CaO:SiO2 ratio. Although the percent chromium leached at any particular CaO:Si2 ratio is different under the two testing conditions (slurry and lysimeter), the pattern of minima and maxima are the same;
i.e., a minimum occurs at about a 1.3 CaO:SiO2 ratio and a maximum at about a 2.0 ratio.
It is also evident from figure 4 that addition of lime to slag C and remelting in air increased the chromium leachability dramatically. For the most part, calcium concentration in the leachate increased in proportion to the lime addition to the slag, but sulfur in the leachate remained relatively constant, reflecting the saturation and precipitation of gypsum. The results indicate that waste chromium-bearing slags are particularly susceptible to leaching of chromium by acid precipitation when they contain calcium in amounts sufficient to give a CaO:SiO2 ratio of about 2.
The effect of lime addition on chromium leachability from waste slag has a counterpart in the well-known process of chemical extraction of chromite (FeO·Cr2O3) ores. In this case, high chromium leachability is desired and is aided by the addition of lime to a mixture of chromite ore and Na2CO3. The CaO-Na2CO3 ore mixture is roasted at 1,100° to 1,150° C in an oxidizing air atmosphere, followed by water leaching of the roasted material to extract the chromium as Na2CrO4 in solution. The extract solution is acidified and purified, and the chromium is recovered by chromic acid electrolysis or by other means. The lime addition increases the ore roasting rate and is believed to inhibit the solubility of undesirable impurities.
The beneficial effect of lime on chromite ore roasting was noted by Doerner more than 50 years ago. Doerner proposed that lime may react with FeO·Cr2O3 to form intermediate CaCrO4, followed by reaction of the chromate with Na2CO3 to form Na2CrO4. He also found that CaO enabled the oxidation of pure Cr2O3 to chromate under air at atmospheric pressure and 900° C. The conversion of Cr2O3 to chromate was determined to be about 70 pct complete in the presence of CaO, while Cr2O3 alone under the same conditions did not oxidize. No mention was found in the chromite-roasting literature regarding an optimum lime addition that was dependent on chromite ore silica content.
Phase Diagram Evidence for CaO:SiO2 Influence
The influence of the CaO:SiO2 ratio on the chromium leachability from waste chromium-bearing slags, particularly the maximum occurring at about 2.0 ratio, is an important result of the present work, which can be explained from a study of phase diagrams from related systems. An examination of the CaO-Cr2O3-SiO2 system delineated by Glasser and Osborn indicates the importance of the 2.0 ratio to waste slag chromium leachability. Figure 5 presents the subsolidus phase diagram for the ternary system at 1,350° C. At the low chromium concentrations (less than 8.1 pct) encountered in the tested waste slags, the diagram shows a phase transformation near the 2.0 CaO:SiO2 ratio depending on the slag’s chromium content. If lime is added to the system in the 1.5 to 2.0 CaO:SiO2 region, two phase boundaries are crossed near the 2.0 ratio and Cr2O3 is converted to CaO·Cr2O3 (calcium chromite).
Oxidation of CaO·Cr2O3
CaO·Cr2O3 not greatly soluble in acid, but in the presence of air and CaO at elevated temperatures it is readily oxidized to acid-soluble CaCrO4 (calcium chromate). The relative insolubility of CaO·Cr2O3 is evident in table 2, where the CaO·Cr2O3 in the base
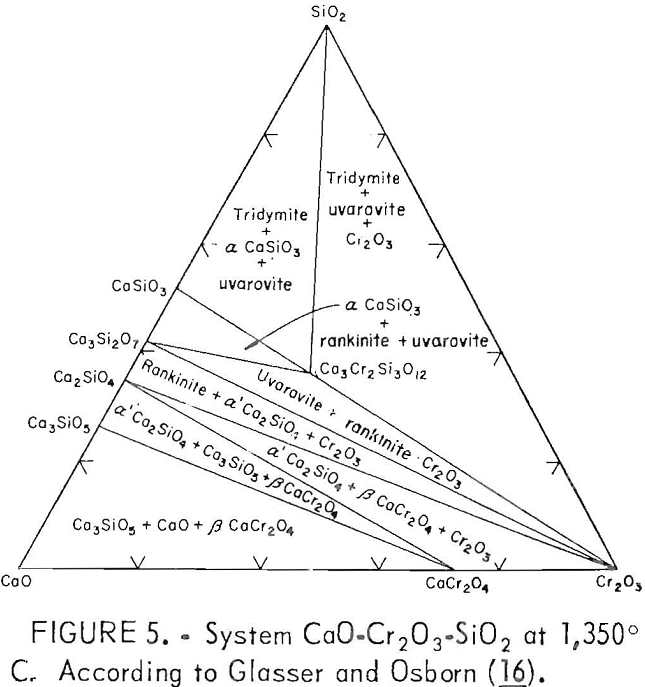
slag A (presumed present, as derived from the phase diagram of figure 5 for a 2.66 CaO:SiO2 ratio) did not leach under the lysimeter test conditions. However, the greater solubility of CaO·Cr2O3 compared with that of other chromium phases below the 2.0 CaO:SiO2 ratio was revealed when the base slags were subjected to the stronger acid and higher liquid-to-solid ratio of the slurry testing (table 3 and fig. 3).
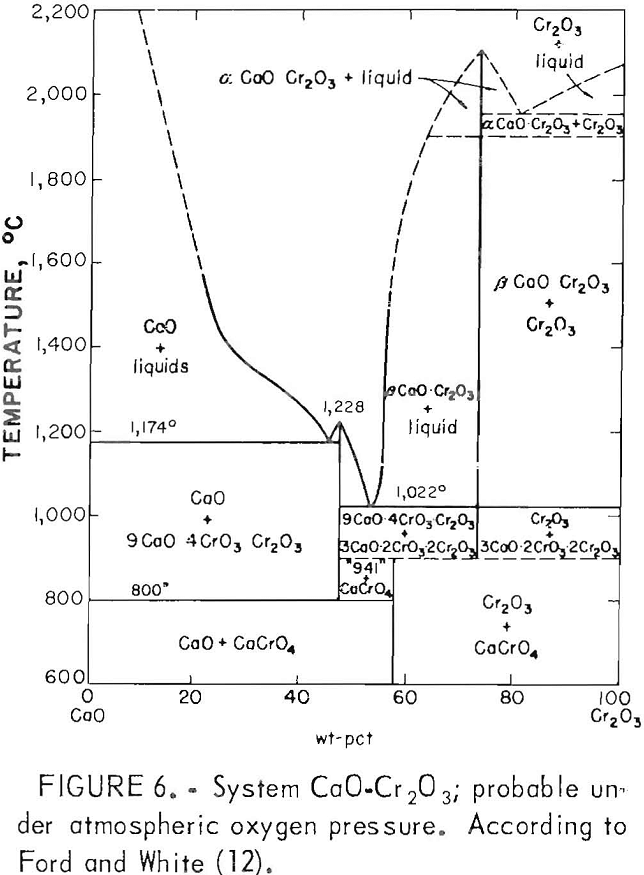
The oxidation of CaO·Cr2O3 to CaCrO4 renders the slag’s chromium content very susceptible to leaching, even under the milder conditions of lysimeter testing (table 2, slags C3 through C5). The preparation of altered slags by melting for an extensive period under air atmosphere ensures the oxidation of CaO·Cr2O3 to CaCrO4, which explains the greater chromium leachability of altered slags in the composition range of CaO·Cr2O3 formation. Doerner believed the CaO·Cr2O3 oxidation might pass through an intermediate stage with formation of the compound 3CaO-CrO3-Cr2O3, followed by final conversion to CaCrO4. But Ford and White’s probable phase diagram (fig. 6) for the CaO-Cr2O3 binary system under atmospheric oxygen pressure indicates that 9CaO-4CrO3·Cr2O3 is a more likely intermediate. The phase diagram shows the likelihood of CaCrO4 forming from CaO·Cr2O3 in waste slag as it is cooled from higher temperatures in air.
It should be pointed out that CaCrO4 cannot exist in the slag while in the steelmaking furnace. Before tapping, the slag is in contact with the molten iron bath, which would reduce CaCrO4 by probable slag-metal reactions such as those shown at bottom of this page (1-2). The poor leachability of base slag A in the lysimeter test compared with that of altered slags of similar CaO:SiO2 ratio probably indicates that slag A had not oxidized appreciably. Apparently when the slag was tapped from the industrial furnace and cooled, time for oxidation of CaO·Cr2O3 was insufficient despite the severance of slag-metal bath contact. Entrained metallic particles in the slag may also inhibit oxidation during this cooling period
The question arises as to whether CaO·Cr2O3 in waste slag would oxidize to yield soluble CaCrO4 under conditions of disposal in the environment (long exposure to air, summer heat, and acid precipitation) . The present work does not answer this question with certainty, owing to the short leaching periods used. The free energy change for the oxidation of CaO·Cr2O3 was not calculated since a free energy value for the compound could not be located in the thermodynamic literature. However, thermodynamic calculations show that the oxidation reaction
2CaO + Cr2O3 + 1.5O2 → 2CaCrO4……………………………………………..(3)
is feasible at ambient temperatures. The standard free energy of this reaction was calculated to be -71.6 kcal. Dufau found experimentally that the oxidation of CaO·Cr2O3 begins below 100° C in air. Therefore, waste slag containing chromium in the form of CaO·Cr2O3 is believed
particularly vulnerable to oxidation to soluble CaCrO4 when exposed for a long period in the environment.
Selection of CaO:SiO2 Ratios for Prevention of Chromium Leaching
A slag composition having a CaO:SiO2 ratio near 2.0 is to be generally avoided, since it predisposes the slag to the formation of oxidizable CaO·Cr2O3. The phase diagram of figure 5 indicates that CaO:SiO2 ratios greater than 2.0 should lead to additional CaO·Cr2O3 formation and, thence, to greater chromium leachability. However, the experimental data do not correspond well with this phase diagram information. The large dropoff in chromium leachability for altered slags having ratios greater than about.
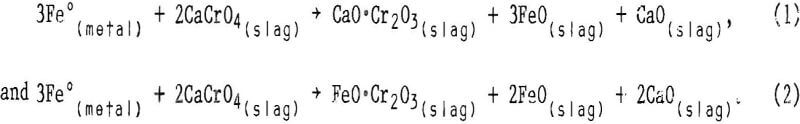
2.6 (fig. 4) is not completely understood. The moderately reduced chromium leachability between the ratios 2.0 and 2.6 is reasonable, since lime addition beyond the maximum point of the 2.0 CaO:SiO2 ratio does not create more CaO·Cr2O3 but simply dilutes the slag and consumes acid. The severe dropoff between 2.6 to 3.4, however, requires a different explanation, which may possibly be in the limitations of the leaching system. High lime addition could cause excessive precipitation and adsorption of gypsum on the slag residue. This adsorbed layer could act as a barrier to prevent reaction between the H2SO4 leachant and CaCrO4 in the slag. Figure 3 indicates the phenomenon is influenced by acid strength. The unaltered slags of figure 3 display no dropoff of chromium leachability at high CaO:SiO2 ratios for the stronger acid concentrations. The result suggests that the gypsum barrier, if it exists, dissolves in the stronger acid to allow normal chromium leaching.
In figures 3 and 4, the chromium leach-ability increases as the CaO:SiO2 ratio is decreased below the range around 1.2 to 1.3, the level of minimum leachability. The appearance of a more leachable chromium phase is evidently occurring. This result complicates the addition of silica proposed in the patent by Nagaya. Optimum suppression of chromium leachability appears to require a consideration of the slag CaO:SiO2 ratio, a factor not considered by Nagaya in his patent.
Influence of MgO
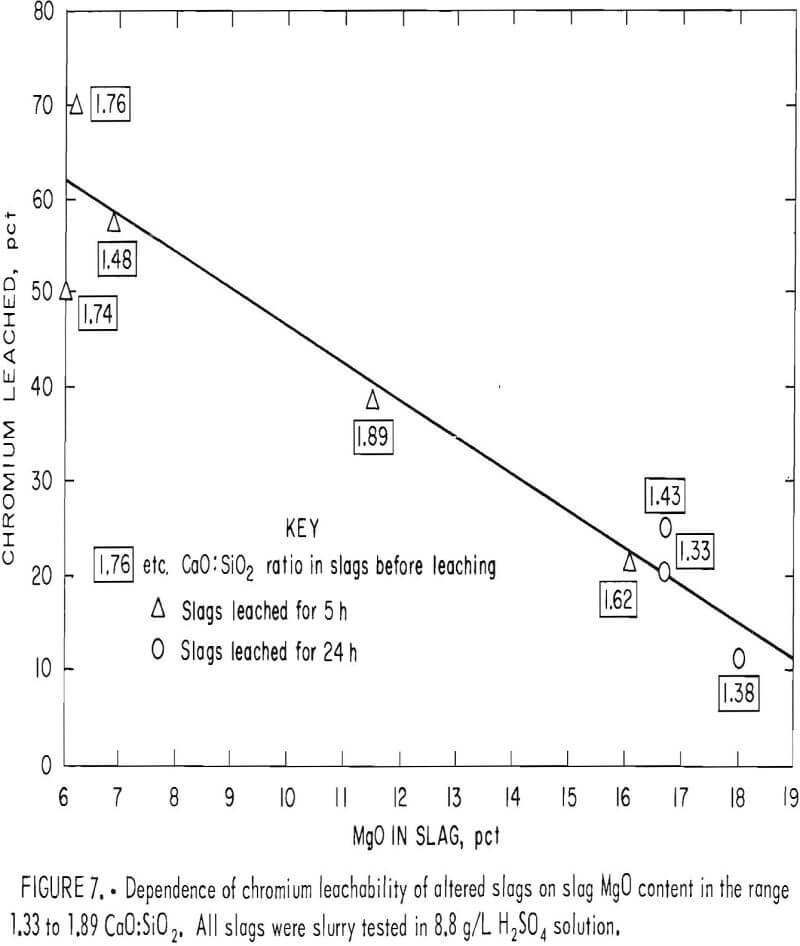
When the chromium leachability of various alterations of slags A and B were compared with those of altered slag C, numerous deviations indicated that the CaO:SiO2 ratio was not the sole composition factor controlling chromium leachability. The variation seemed random for slags having CaO:SiO2 ratios higher than 1.9, but in the region between ratios of 1.2 and 1.9 the leachability appeared to follow a sequence related to slag magnesium concentration. In this region, chromium leached less readily as the magnesium content in the slag was increased. This result is illustrated in figure 7, which plots the chromium leachability of altered slags in slurry testing (slags A2 through A4, B1 through B3, and C6 and C7) against the slag MgO content in the region of interest. The chromium leachability as a function of magnesium concentration appears to have approximately linear characteristics in the 6- to 20- pct-MgO range examined. The dependence of chromium leachability on magnesium content was further supported by a similar plot (fig. 8) of the slurry test data from base slags A, B, C, and D. As in figure 3, the percentages of leached chromium for slags A and C are interpolated values. Figure 8 shows that increasing the magnesium content in slag again depresses the slag chromium leachability, with the exception of slag A at the stronger acid concentrations. In this case, the high CaO:SiO2 ratio (2.66) is the dominating factor influencing chromium leachability rather than slag MgO content.
Formation of MgO·Cr2O3
The inhibiting effect of increased slag magnesium content on chromium leachability, in the range below a 2.0 CaO:SiO2 ratio, appears to originate from the formation of insoluble spinel compounds. White’s research into refractory-slag systems reports a fact remarkably pertinent to waste slag chromium leachability. He showed that the CaO:SiO2 ratio of 2.0 was a critical factor in determining the phase combinations present in chrome-magnesite refractory. White
determined that the phase combinations up to a ratio of 2.0 CaO:SiO2 contained all the chromium in a spinel, solid-solution phase with MgO, FeO, Fe2O3, and Al2O3. At ratios greater than 2.0, the spinel structure gave way to formation of calcium chromites, ferrites, and aluminates.
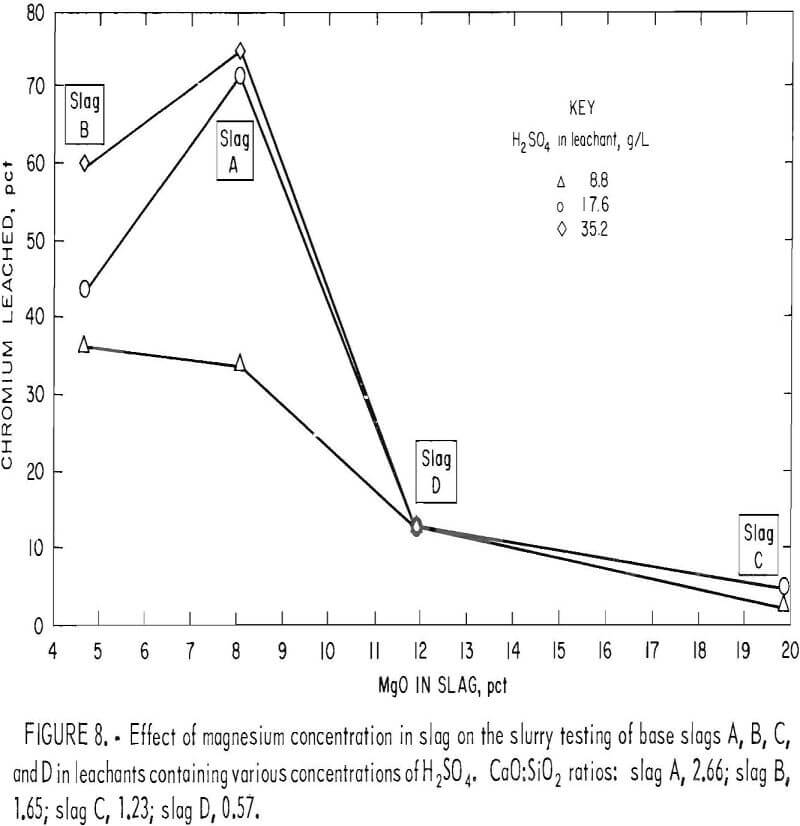
The quaternary phase diagram for the system CaO·MgO·Cr2O3·SiO2, shown in figure 9, was developed by Berezhnoi and Gul’ko. Examination of this phase diagram indicates that formation of the MgO·Cr2O3 spinel (picrochromite) is likely responsible for the diminished chromium leachability for slags of high magnesium content. As the tetrahedron of the system is traversed through its internal elementary tetrahedra enclosing various phase domains, the compositional requirements for existence of MgO·Cr2O3 become evident. Thus, if a traverse is made in the direction of increasing lime content at relatively low and constant Cr2O3 and MgO levels (indicated by the arrow on figure 9), the following domains will be traversed:

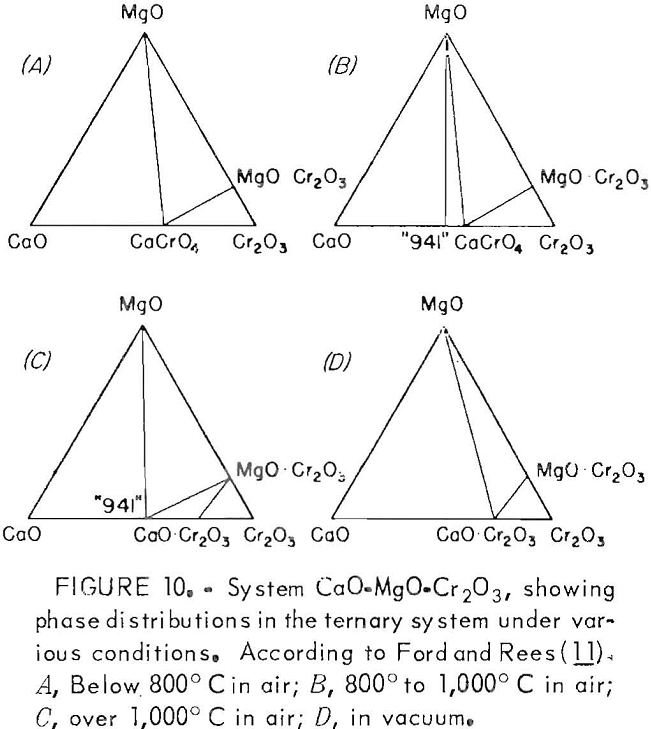
It is evident that the formation of MgO·Cr2O3 spinel does not occur after crossing the phase boundary slightly beyond the 2.0 CaO:SiO2 ratio. Above the 2.0 ratio, chromium exists wholly in the CaO·Cr2O3 state, which, as determined earlier, is readily oxidized to acid- soluble CaCrO4 under appropriate conditions. Ford and Rees developed a similar diagram for the system, but they made their phase determinations in an air atmosphere, with resulting partial oxidation of CaO·Cr2O3 occurring as a consequence.
Leaching and Oxidation Resistance of MgO·Cr2O3
MgO·Cr2O3 is an exceedingly stable compound that is very resistant to dissolution by mineral acids. Dufau observed that the compound was attacked with difficulty by HF and HCl, while Ebelman said it was not attacked at all by concentrated HCl. Doerner found MgO·Cr2O3 to be insoluble even in HNO3. Figure 8 likely reveals the superior resistance of MgO·Cr2O3 toward acid leaching compared with other chromium species in the slag, such as Cr2O3 , FeO·Cr2O3, CaO·Cr2O3, and mixed spinel (Mg, Fe, Al)O·Cr2O3. The greater leachability of base slag A (fig. 8) probably demonstrates the greater solubility of CaO·Cr2O3 in strong H2SO4 solution compared with that of the spinel and other chromium compounds present in slags B, C, and D.
The MgO·Cr2O3, unlike CaO·Cr2O3, is very resistant to oxidation. Bayer and Wiedemann studied the reaction of MgO with Cr2O3 in air using differential thermal analysis,, They found the stable spinel MgO·Cr2O3 was formed preferentially. Oxidation to soluble magnesium chromate (MgCrO4) required high oxygen pressures. This resistance of MgO-Cr2O3 to oxidation under normal conditions was confirmed by Ford and Rees.
Figure 10 gives the ternary phase diagrams they developed for the system CaO·MgO·Cr2O3 under various temperatures in air (A, B, and C) and under vacuum (D). The diagrams show the susceptibility of the system to form oxidation products from CaO·Cr2O3 (CaCrO4 and 9CaO·4CrO3·Cr2O3) in air at elevated temperature. In contrast, MgO·Cr2O3 remains unaffected, whether in air or vacuum. Therefore the MgO·Cr2O3 is a most desirable form for chromium to be disposed to the environment since it possesses excellent resistance both to oxidation and solubility under conditions of acid precipitation.
Ford and Rees (10) also reported that the system CaO·SiO2·Cr2O3-MgO is analogous to the systems CaO·SiO2·Cr2O3·Fe2O3 and CaO·SiO2·Cr2O3·Al2O3. The oxidation product, CaCrO4 , would again be expected to form in the systems when the CaO:SiO2 ratio achieved a 2.0 value.
Therefore, in more complex actual slag systems containing both iron oxide and alumina, the chromium leachability may still be expected to increase remarkably near the 2.0 CaO:SiO2 ratio.
The normally high calcium and low magnesium content of steelmaking slags from the BOF may be the primary cause of the high chromium concentrations found in ground water near a steel mill landfill noted earlier. A CaO:SiO2 ratio of 2.0 to 3.0 is desired in BOF slags for impurity removal, while MgO content is generally less than 6 pct owing to the MgO saturation limit for this type of slag. Generally, BOF slag is disposed as waste rather than recycled or used for construct-ion purposes. Even though the average chromium content of BOF slags runs less than 1.0 pct, the high CaO:SiO2 ratio and low MgO content may allow sufficient leaching of chromium from these slags to contaminate ground water.
Fixing of Chromium
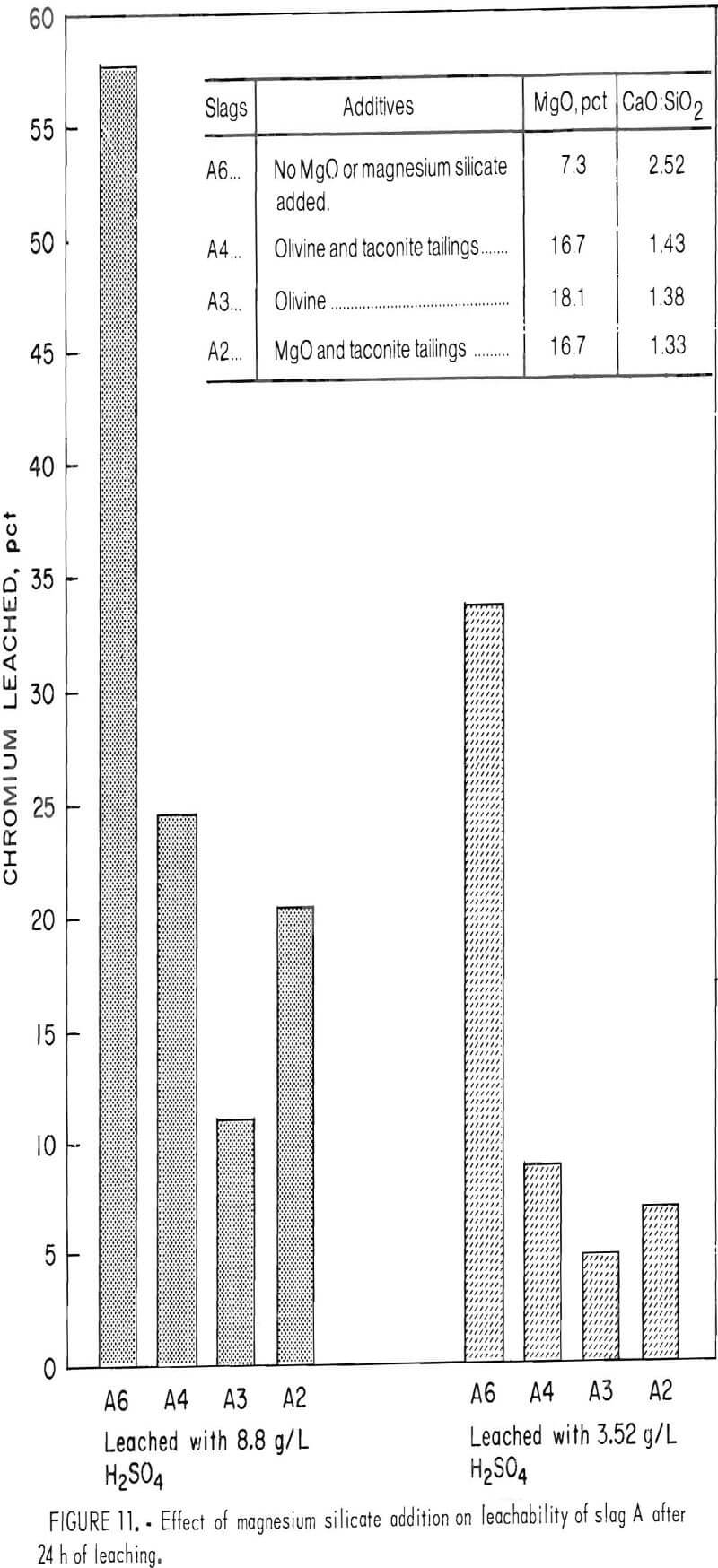
Slags A2, A3, A4, and A6 in tables 1 and 3 represent a set of experiments performed to test the practicality of adding inexpensive magnesium silicate materials
to molten waste slag in order to promote resistance to chromium leaching after the slags are disposed in the environment. Taconite tailings and olivine [2(Mg,Fe)O·SiO2] were used in the tests, but any material such as waste asbestos and discarded magnesite (a common furnace refractory lining used in steelmaking) containing mostly silicate and/or magnesium should be suitable.
The strategy proposed for fixing chromium is to add silica or silicate material to lower the CaO:SiO2 ratio to about 1.3 to 1.4, followed by magnesium addition to maximize the chromium fixation in the form of the stable, insoluble MgO·Cr2O3 spinel. The additions would preferably be made to the charge before the smelting of the chrome ore or ferro-chrome alloy if such a procedure is compatible with the smelting process. This would ensure that the slag phases had reached equilibrium before disposal. If the altered slag composition is not compatible with a particular smelting process, then the additives could be added as powder to the molten slag stream as it is tapped from the furnace at the conclusion of smelting. However, the reaction time to form insoluble chromium species would be shorter for this treatment method.
Slag A was selected for treatment in the tests. This slag showed high chromium leachability in the previous slurry testing and thus was a logical candidate for testing the chromium-fixing scheme. Slag A6 is essentially the same as unaltered slag A except that it was calcined and melted so as to be more comparable to treated slags A2, A3, and A4. CaF2 and Fe2O3 also had to be added to slag A to permit pouring this very viscous slag from the platinum-rhodium crucible at the end of the melting period. Slag A was altered with additions of MgO, taconite tailings, and olivine to obtain the target compositions of about a 1.4 CaO:SiO2 ratio and 16 to 19 pct MgO (slags A2, A3, A4). All of the slags were calcined and melted according to the conditions described earlier. The slags were slurry leached for 24 h with 8.8- and 3.52- g/L-H2SO4 leachant, as shown in table 3.
The results displayed in table 3 and in the bar graph of figure 11 demonstrate the feasibility of fixing chromium in waste slag through addition of cheap magnesium silicate materials. Olivine addition alone (slag A3) improved the resistance of slag A to chromium leachability 81 pct when leached with 8.8 g/L H2SO4 and 85 pct when leached with 3.52- g/L-H2SO4 solution.
Conclusions
It is concluded that certain compositions of chromium-bearing waste slag are susceptible to leaching of their chromium content into the environment under conditions of acid precipitation. There are two composition factors that predispose slag to chromium leachability: (1) the CaO:SiO2 ratio and (2) the magnesium content. When the ratio is above 2.0, chromium is present in normal ferrous slags (where CaO and SiO2 are the majority constituents) as calcium chromite (CaO·Cr2O3), regardless of the magnesium content. CaO·Cr2O3 is slightly soluble in acid and might leach significantly under severe acid precipitation conditions. Of more concern is the possibility that CaO·Cr2O3 is likely to oxidize to calcium chromate (CaCrO4) under conditions of disposal in the environment. CaCrO4 would be readily soluble when exposed to acid precipitation in the industrialized areas of the United States where slags are normally discarded. The chromium leached would be in the hexavalent state, the condition most dangerous to humans and vegetation. This potential problem can be minimized if the slag composition is well below a 2.0 CaO:SiO2 ratio, with enough magnesium present to form the in-soluble picrochromite (MgO·Cr2O3).
It is recommended that leaching of chromium from chromium-bearing waste slag be prevented by adding minerals or other substances containing magnesium and silicate to the slag prior to disposal. The substances would be added to the charge before smelting (if compatible with smelting process) or to the molten slag at time of tapping from the industrial steelmaking furnace. Magnesium silicates are available, inexpensive, and abundant for this purpose. Some magnesium silicates, such as waste asbestos or taconite tailings, are disposal problems in themselves. Their use in treating waste slags could help mitigate two waste disposal problems simultaneously. Abundant minerals that might be used to fix chromium in slag as the spinel MgO·Cr2O3 are olivine, serpentine, some of the micas, and possibly waste tailings from metallurgical beneficiation operations. Discarded, used magnesite refractories are available around any foundry operation and could be used in combination with sand or tailings to provide the magnesium and silica needed to bring the slag composition below the 2.0 CaO:SiO2 ratio to form MgO·Cr2O3.